Phosphovimentin appeared as one of the proteins associated with biotinylated Mepiroxol plasma membrane-bound proteins in 5HT-stimulated platelets. SERT could be one of the other phosphovimentin-associated membrane proteins, but our co-IP data in 5HT-stimulated platelets also demonstrated an elevation in the association of SERTphosphovimentin in whole platelet. Therefore, we tested SERT-phosphovimentin association in 5HT-stimulated platelets. The effects of 5HT-stimulation on the amount of intracellular SERT mirrored those of the cell surface SERT. Previously, it has been shown that 5HT-stimulation phosphorylates vimentin on the Serine56 residue, but the vimentin S56A mutant is not phosphorylated by 5HT-stimulation. Therefore, to mechanistically determine how the vimentin-SERT association responds to 5HT for regulating the distribution of transporter molecules between plasma membrane and intracellular locations, the S56A mutant and the C-terminus truncated forms of SERT were studied in a CHO heterologous expression system. Obviously, not all aspects of 5HTbiology in platelets can be recapitulated in CHO cells, but the CHO model system allows for the analysis of the association between vimentin and the C-terminus truncated forms of SERT and the nonphosphorylated mutant form of vimentin S56A. To ascertain 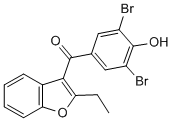 the optimal 5HT concentration required to stimulate CHO cells expressing hSERT, we measured the density of SERT proteins on the plasma membrane of CHO-hSERT cells and compared this finding to human platelet membranes using biotinylation. In this previous study, we tried to model the effect of plasma 5HT on platelet SERT in a heterologous expression system. Simply stated, an equal amount of biotinylated membrane proteins from CHO-SERT cells and platelets were resolved and analyzed by W/B using SERT-Ab. These calculations together with the dose response analysis of CHO cells to 5HT-stimulation which was already conducted in our previous studies showed that the expression of SERT on the plasma membrane of CHO-SERT cells was 59-fold higher than on the platelet membrane. This estimation indicated that the effect of plasma 5HT at a concentration of 1 nM on platelet SERT may Dexrazoxane hydrochloride correspond to exogenous 5HT at a concentration of,45 mM on CHO-SERT cells. The co-localization of the red vimentin and green YFP-SERT signals were captured in the overlaid images with YFP and Texas Red filter sets.
the optimal 5HT concentration required to stimulate CHO cells expressing hSERT, we measured the density of SERT proteins on the plasma membrane of CHO-hSERT cells and compared this finding to human platelet membranes using biotinylation. In this previous study, we tried to model the effect of plasma 5HT on platelet SERT in a heterologous expression system. Simply stated, an equal amount of biotinylated membrane proteins from CHO-SERT cells and platelets were resolved and analyzed by W/B using SERT-Ab. These calculations together with the dose response analysis of CHO cells to 5HT-stimulation which was already conducted in our previous studies showed that the expression of SERT on the plasma membrane of CHO-SERT cells was 59-fold higher than on the platelet membrane. This estimation indicated that the effect of plasma 5HT at a concentration of 1 nM on platelet SERT may Dexrazoxane hydrochloride correspond to exogenous 5HT at a concentration of,45 mM on CHO-SERT cells. The co-localization of the red vimentin and green YFP-SERT signals were captured in the overlaid images with YFP and Texas Red filter sets.
If vimentin and SERT localized then the structures would subjected to immunoblot analysis with pS56-Ab
Leave a reply
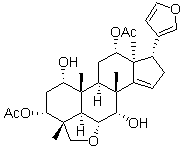 becomes a bridge between vimentin and the plasma membrane. At physiological plasma 5HT levels, vimentin-SERT association was found at intracellular locations and on the plasma membrane. However, when plasma 5HT level was higher than physiological level, their association was enhanced and the level of SERT on the plasma membrane was decreased. Therefore, we hypothesize that SERT utilizes the vimentin network during translocation from the plasma membrane. Furthermore, the 5HT-dependent phosphorylation of vimentin on the S56 residue accelerates the translocation of SERT on the 5HT-altered vimentin network. Future analysis of these mutants in stable transfection systems, as well as continued experiments with the phospho-mimicking mutants presented here, will further reveal the mechanism of action that governs transporter C-terminal phosphorylation.
becomes a bridge between vimentin and the plasma membrane. At physiological plasma 5HT levels, vimentin-SERT association was found at intracellular locations and on the plasma membrane. However, when plasma 5HT level was higher than physiological level, their association was enhanced and the level of SERT on the plasma membrane was decreased. Therefore, we hypothesize that SERT utilizes the vimentin network during translocation from the plasma membrane. Furthermore, the 5HT-dependent phosphorylation of vimentin on the S56 residue accelerates the translocation of SERT on the 5HT-altered vimentin network. Future analysis of these mutants in stable transfection systems, as well as continued experiments with the phospho-mimicking mutants presented here, will further reveal the mechanism of action that governs transporter C-terminal phosphorylation. this comparative analysis allows us to contrast ICU patients with models of muscle atrophy, muscle inflammatory and muscle inactivity along the lines presented by Sacheck et al. It also facilitates discussion of the utility of such models and whether they accurately represent human muscle wasting disorders. In two recent publications, common genes up regulated or down regulated in animal models with muscle wasting have been identified. In the first study animal models for fasting, cancer cachexia, uremia and diabetes mellitus identified 120 unique genes involved in catabolism, and the authors named these ‘atrogens’. In a second paper these genes were
this comparative analysis allows us to contrast ICU patients with models of muscle atrophy, muscle inflammatory and muscle inactivity along the lines presented by Sacheck et al. It also facilitates discussion of the utility of such models and whether they accurately represent human muscle wasting disorders. In two recent publications, common genes up regulated or down regulated in animal models with muscle wasting have been identified. In the first study animal models for fasting, cancer cachexia, uremia and diabetes mellitus identified 120 unique genes involved in catabolism, and the authors named these ‘atrogens’. In a second paper these genes were  the aggregation of mutant huntingtin or ataxin-3 proceeds by nucleated growth polymerization into protein fibrils that structurally resemble amyloid fibrils and react with amyloid-specific histochemical dyes such as Congo Red or thioflavin. Analogous biophysical properties are observed for b-amyloid and a-synuclein fibrils associated with Alzheimer��s and Parkinson��s diseases, respectively. Amyloid fibrils are thought to be nucleated by monomers or globular oligomers of misfolded protein. In turn, fibrils may co-assemble into much larger insoluble protein aggregates that are resistant to proteolysis. While it is clear that protein misfolding can elicit cellular toxicity, whether fibrillar protein aggregates are
the aggregation of mutant huntingtin or ataxin-3 proceeds by nucleated growth polymerization into protein fibrils that structurally resemble amyloid fibrils and react with amyloid-specific histochemical dyes such as Congo Red or thioflavin. Analogous biophysical properties are observed for b-amyloid and a-synuclein fibrils associated with Alzheimer��s and Parkinson��s diseases, respectively. Amyloid fibrils are thought to be nucleated by monomers or globular oligomers of misfolded protein. In turn, fibrils may co-assemble into much larger insoluble protein aggregates that are resistant to proteolysis. While it is clear that protein misfolding can elicit cellular toxicity, whether fibrillar protein aggregates are  of the transporter since our results indicate that the triple mutation DDD of 611, 613, and 616 retained only 5% of its 5HT uptake capacity as compared to control SERT. It is also important to note that the presence of such a large amount of negative charge on the end of the protein could cause alterations in protein folding or proteinprotein associations that are important for protein function, resulting in the observed blunting of transport capacity. Next, we analyzed the impact of four truncations of the SERT C-terminus on the trafficking and expression of SERT on the plasma membrane using biotinylation and IF assays. Our data indicate that depending on the amount of truncation from the Cterminus of SERT, there was altered localization of the transporter. Therefore, we carried out a biotinylation analysis on some of the phosphorylation-mimicking mutations in an effort to determine the plasma membrane localization of these mutants, i.e., whether the mutation arrests them intracellularly or whether the mutants can still traffic to the plasma membrane. The data indicate that 3 possible phosphorylation sites do contribute to the 5HT uptake rates of transporters via inhibiting their proceedings toward the plasma membrane.
of the transporter since our results indicate that the triple mutation DDD of 611, 613, and 616 retained only 5% of its 5HT uptake capacity as compared to control SERT. It is also important to note that the presence of such a large amount of negative charge on the end of the protein could cause alterations in protein folding or proteinprotein associations that are important for protein function, resulting in the observed blunting of transport capacity. Next, we analyzed the impact of four truncations of the SERT C-terminus on the trafficking and expression of SERT on the plasma membrane using biotinylation and IF assays. Our data indicate that depending on the amount of truncation from the Cterminus of SERT, there was altered localization of the transporter. Therefore, we carried out a biotinylation analysis on some of the phosphorylation-mimicking mutations in an effort to determine the plasma membrane localization of these mutants, i.e., whether the mutation arrests them intracellularly or whether the mutants can still traffic to the plasma membrane. The data indicate that 3 possible phosphorylation sites do contribute to the 5HT uptake rates of transporters via inhibiting their proceedings toward the plasma membrane.