Given the enormous implications in regulation of cellular function by the modulation of gene expression that regulate gene expression. Transcription regulatory machinenary in Folinic acid calcium salt pentahydrate eukaryotes involves specific transcription factors and transcription inhibitors; proteins that are required to turn on and turn off the expression of particular genes. These considerations point to a possible mechanism of how oxalate differentially regulates the gene expression of such a large number of genes. Given that oxalate is a metabolic end product in humans that cannot be further metabolized, such large scale changes in gene expression in renal epithelial cells in response to high oxalate levels points to an indirect mechanism of action, which may involve the interaction of oxalate with the cell membrane or in intracellular components. The primary site of oxalate action in cell remains unknown. Irrespective of primary site of action, one of the most common means by which cells sense changes is by activating the signal transduction pathways, especially the stress signal pathways. The stress associated signals are transduced through a series of proteins that are activated by phosphorylation/dephosphorylation steps and are finally turned into transcription factors, causing changes in gene expression. Though the present study design does not allow for the identification of activity changes due to phosphorylation, we identified changes in the gene expressions of upstream activators of several signaling pathways. Proteins like Ras, Fas and MKK are highly up-regulated as a result of oxalate exposure. These proteins are known to play important roles in JNK/SAPK signaling and p38 MAPK signaling. These results are in agreement with previous studies, by us and others, that identified an active role for Stress Activated Protein Kinases in oxalate renal cell interactions. We also identified changes in the expression of genes associated with retinoic Acid Receptor Signaling Pathway. Clearly additional studies are required to evaluate the functional consequence of these gene expression changes. In summary, our study is the first attempt at profiling the Genome-wide expression changes in human renal  epithelial cells as a result of exposure to oxalate. Results from our study point to complex and intricate mechanisms, including differential gene expression, in renal epithelial cells in response to oxalate exposure. Clearly further studies are required to completely understand the implications of the plethora of changes in gene expression occurring as a result of oxalate exposure in renal epithelial cells. We must separate and characterize the genes that are derived from the by-stander effect and identify the genes whose altered expression is responsible for oxalate nephrotoxicity. Malaria due to Plasmodium falciparum remains a major global health burden and a leading cause of death worldwide among children under five. Benzethonium Chloride Increasing drug resistance, including emerging resistance to the artemisinin drugs, and the declining efficacy of vector control interventions in some populations make the development of effective malaria vaccines an urgent priority. During blood-stage infection, P. falciparum merozoites invade erythrocytes, mediated by the release of invasion ligands from apical organelles that interact with receptors on the erythrocyte surface. The repertoire of invasion ligands includes two major families, the P. falciparum reticulocyte-binding homologues, and erythrocyte binding antigens. The ability of P. falciparum to vary the expression and/or use of EBA and PfRh proteins enables the use of alternate invasion pathways, facilitating immune evasion that enables P. falciparum to cause repeated and chronic infections. Invasion pathways can be broadly classified into two main pathways, sialic acid dependent invasion and SA-independent invasion.
epithelial cells as a result of exposure to oxalate. Results from our study point to complex and intricate mechanisms, including differential gene expression, in renal epithelial cells in response to oxalate exposure. Clearly further studies are required to completely understand the implications of the plethora of changes in gene expression occurring as a result of oxalate exposure in renal epithelial cells. We must separate and characterize the genes that are derived from the by-stander effect and identify the genes whose altered expression is responsible for oxalate nephrotoxicity. Malaria due to Plasmodium falciparum remains a major global health burden and a leading cause of death worldwide among children under five. Benzethonium Chloride Increasing drug resistance, including emerging resistance to the artemisinin drugs, and the declining efficacy of vector control interventions in some populations make the development of effective malaria vaccines an urgent priority. During blood-stage infection, P. falciparum merozoites invade erythrocytes, mediated by the release of invasion ligands from apical organelles that interact with receptors on the erythrocyte surface. The repertoire of invasion ligands includes two major families, the P. falciparum reticulocyte-binding homologues, and erythrocyte binding antigens. The ability of P. falciparum to vary the expression and/or use of EBA and PfRh proteins enables the use of alternate invasion pathways, facilitating immune evasion that enables P. falciparum to cause repeated and chronic infections. Invasion pathways can be broadly classified into two main pathways, sialic acid dependent invasion and SA-independent invasion.
The PfRh ligands are located in the rhoptries of merozoites and specific mechanisms are in place in eukaryotic cells
Leave a reply
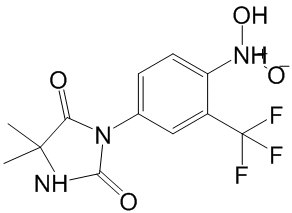
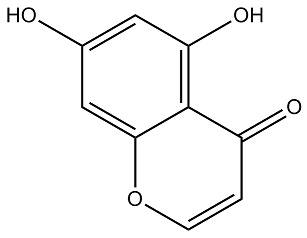 mechanisms falls outside the scope of our work, although it is tempting to speculate that transcription factors binding in proximity of HIFs may be involved in recruitment of co-activator or co-repressor proteins. Of note, a recent mammalian two-hybrid survey of protein-protein interactions for human and mouse TFs reported a physical association between HIF1A and AP-1 family member JUN, as well as the previously known interaction between CEBPB and p300. p300/CBP is a master co-activator of HIF-mediated transcription whose recruitment can also be mediated by CREB. In this regard, evidence from a synthetic transactivation screen on the EGLN1 promoter pointed to ETV4 as an additional p300-dependent coactivator of HIF-mediated transcription. Moreover, HIF1 is known to interact with Jab1/CSN5, a protein originally identified as a transcriptional coactivator for AP1. Future investigations on protein composition of HIF-bound enhancers should be pivotal in supporting this model. The associations between HIFs and AP1, CREB and CEBPs analyzed in our reporter results could be general across many HIF targets or be restricted to individual targets. To judge the generality of these results, we conducted a gene-set enrichment analysis of transcription factor targets in a sorted list of genes regulated by hypoxia. The results of this analysis showed a significant enrichment of CEBP targets among hypoxiainducible genes, suggesting that at least for this family of transcription factors, the functional association with HIFs could be relatively general. Of note, recent works have reported a direct protein-protein interaction between HIF1a and CEBPa, and have implicated CEBPa activity in regulation of the HIF target genes galectin-1 and PAI-1. Hypoxic induction of both galectin-1 and PAI-1 was found to be synergistically dependent on both HIF1a and CEBPa activity and their co-binding to the promoter region. Our results further suggest that this functional association may be general across a wider collection of HIF targets. In conclusion, the data presented herein demonstrates that integration of high-throughput chromatin immunoprecipitation and gene expression data is a successful approach to select highquality core HIF binding regions, and provides experimental proof of principle for the biological relevance of enriched transcription factor binding sites other than the HIF binding consensus in HIFmediated transcription. Specifically, our results suggest that
mechanisms falls outside the scope of our work, although it is tempting to speculate that transcription factors binding in proximity of HIFs may be involved in recruitment of co-activator or co-repressor proteins. Of note, a recent mammalian two-hybrid survey of protein-protein interactions for human and mouse TFs reported a physical association between HIF1A and AP-1 family member JUN, as well as the previously known interaction between CEBPB and p300. p300/CBP is a master co-activator of HIF-mediated transcription whose recruitment can also be mediated by CREB. In this regard, evidence from a synthetic transactivation screen on the EGLN1 promoter pointed to ETV4 as an additional p300-dependent coactivator of HIF-mediated transcription. Moreover, HIF1 is known to interact with Jab1/CSN5, a protein originally identified as a transcriptional coactivator for AP1. Future investigations on protein composition of HIF-bound enhancers should be pivotal in supporting this model. The associations between HIFs and AP1, CREB and CEBPs analyzed in our reporter results could be general across many HIF targets or be restricted to individual targets. To judge the generality of these results, we conducted a gene-set enrichment analysis of transcription factor targets in a sorted list of genes regulated by hypoxia. The results of this analysis showed a significant enrichment of CEBP targets among hypoxiainducible genes, suggesting that at least for this family of transcription factors, the functional association with HIFs could be relatively general. Of note, recent works have reported a direct protein-protein interaction between HIF1a and CEBPa, and have implicated CEBPa activity in regulation of the HIF target genes galectin-1 and PAI-1. Hypoxic induction of both galectin-1 and PAI-1 was found to be synergistically dependent on both HIF1a and CEBPa activity and their co-binding to the promoter region. Our results further suggest that this functional association may be general across a wider collection of HIF targets. In conclusion, the data presented herein demonstrates that integration of high-throughput chromatin immunoprecipitation and gene expression data is a successful approach to select highquality core HIF binding regions, and provides experimental proof of principle for the biological relevance of enriched transcription factor binding sites other than the HIF binding consensus in HIFmediated transcription. Specifically, our results suggest that 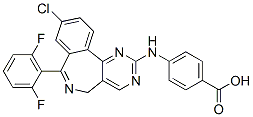 stage and sex. We employed a custom oligonucleotide microarray containing 31,918 ESTs
stage and sex. We employed a custom oligonucleotide microarray containing 31,918 ESTs