From a therapeutic perspective, it is interesting that ISA27 in combination with the conventional chemotherapy drug TMZ inhibited U87MG cell growth. This combination worked in a synergistic manner as confirmed by isobolographic GDC-0199 Bcl-2 inhibitor analysis. This result suggests the possibility of lowering the dose of TMZ used in the treatment of GBM. In conclusion, our data show that ISA27 disrupts the MDM2p53 interaction and releases the powerful antitumor capacities of p53 in GBM cells. The use of this MDM2 inhibitor could offer a novel therapy for the treatment of GBM patients by inhibiting tumor growth. Proteases catalyze the hydrolysis of peptide bonds in proteins and are involved in digestive as well as regulatory processes. In the human genome, approximately 2% of the genes code for proteases. While most proteases are soluble, a small fraction is membrane-embedded. These intramembrane proteases differ from soluble proteases in a variety of aspects: They are composed of a number of transmembrane domains which harbor the catalytic residues with their active sites buried several A ? into the membrane. Their substrates are transmembrane proteins that reside inside the membrane in a dormant form. Upon cleavage, most substrates release a soluble part into the cytosol or extracellular space. It is therefore not surprising that intramembrane proteases are involved in various signaling pathways. There are three families of intramembrane proteases, classified according to their catalytic mechanism: intramembrane metalloproteases, intramembrane aspartic proteases, and intramembrane serine proteases. The latter belong to the family of rhomboid proteins, containing active intramembrane proteases and inactive homologs. Rhomboids are found in all kingdoms of life, but are functionally diverse. They take part in various distinct cellular processes such as the EGFR-signaling pathway in the fruit fly Drosophila melanogaster, quorum sensing in the Gram negative bacterium Providencia stuartii and host cell infection by apicomplexan parasites. Structurally, rhomboids are the best characterized intramembrane proteases. Several different crystal forms of the E. coli rhomboid GlpG have provided insight into the mechanism of intramembrane proteolysis. However, a detailed picture of the rhomboid-substrate interaction is not available. As an alternative, crystal structures of covalent inhibitors bound to GlpG have revealed which areas and residues may play a role in primed and non-primed site interaction, and oxyanion stabilization. The availability of inhibitors is also important for future functional studies. Moreover, potent and BAY 73-4506 selective inhibitors may serve as lead structures for future drug design. Up to date, rhomboid inhibitors have been reported based on three distinct scaffolds: 4-chloro-isocoumarins, fluorophosphonates, and N-sulfonylated beta-lactams. However, these are not selective enough to inhibit only rhomboids within the entire proteome. In addition, these inhibitors are also not promiscuous enough to inhibit rhomboids from different organisms 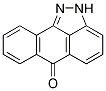 equally well. Therefore, it is still of great interest to find new types of inhibitors. In order to facilitate this search, various screening methods have been employed so far. All of these have relied on monitoring the cleavage of a substrate through gel-based, FRET or MALDI mass spectrometry techniques. However, a limitation of these methods is the availability of a matching protein or polypeptide substrate. Rhomboids from one species may cleave substrates from another species, but this is not a general rule. We therefore reasoned that it would be beneficial to develop an inhibitor assay for rhomboid proteases that does not rely on a substrate at all. A few years ago Cravatt and co-workers developed a highthroughput inhibitor screening method that uses fluorescent activity-based probes. ABPs are small molecules that covalently bind to the active form of an enzyme, but not to an inactivated or zymogen form. ABPs generally consist of a tag, a spacer and an electrophilic group that traps an active site nucleophile. The binding event can be detected by a variety of techniques, such as gel-scanning, biotin blot or fluorescent microscopy, depending on the tagging moiety. When appended to a fluorescent dye, the binding of an ABP can be detected by fluorescence polarization. This so-called fluorescence polarization activity-based protein profiling has been used in inhibitor high-throughput screens for a variety of poorly characterized enzymes.
equally well. Therefore, it is still of great interest to find new types of inhibitors. In order to facilitate this search, various screening methods have been employed so far. All of these have relied on monitoring the cleavage of a substrate through gel-based, FRET or MALDI mass spectrometry techniques. However, a limitation of these methods is the availability of a matching protein or polypeptide substrate. Rhomboids from one species may cleave substrates from another species, but this is not a general rule. We therefore reasoned that it would be beneficial to develop an inhibitor assay for rhomboid proteases that does not rely on a substrate at all. A few years ago Cravatt and co-workers developed a highthroughput inhibitor screening method that uses fluorescent activity-based probes. ABPs are small molecules that covalently bind to the active form of an enzyme, but not to an inactivated or zymogen form. ABPs generally consist of a tag, a spacer and an electrophilic group that traps an active site nucleophile. The binding event can be detected by a variety of techniques, such as gel-scanning, biotin blot or fluorescent microscopy, depending on the tagging moiety. When appended to a fluorescent dye, the binding of an ABP can be detected by fluorescence polarization. This so-called fluorescence polarization activity-based protein profiling has been used in inhibitor high-throughput screens for a variety of poorly characterized enzymes.
the high accumulation of p53 renders the cells highly susceptible to p53 reactivation and more sensitive to apoptosis
Leave a reply
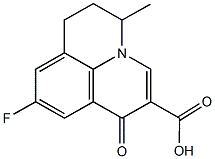 and in the case of lysosomes ‘lysosomotropism’. This accumulation of desipramine 168 results in detachment of the ASM from the inner lysosomal membraneand its subsequent inactivation, probably by proteolytic degradation. Weak bases, therefore, do not directly inhibit ASM, but result in a functional inhibition of ASM. We have thus proposed the acronym FIASMA for Functional Inhibitor of Acid SphingoMyelinAse. According to this model, functional inhibition of ASM requires high lysosomal concentrations of a weak basic drug. Previously, we have shown that functional inhibition of ASM is related to high pKa- and high logP-values and have characterized several new FIASMAs, including the antidepressant drugs doxepine 63, fluoxetine 104, maprotilin 109, nortriptyline 114, paroxetine 118 and sertraline 124. The aims of the present study wereto identify more FIASMAs,to further improve the in silico prediction of functional ASM inhibition by developing compact and easily-interpretable models with high internal
and in the case of lysosomes ‘lysosomotropism’. This accumulation of desipramine 168 results in detachment of the ASM from the inner lysosomal membraneand its subsequent inactivation, probably by proteolytic degradation. Weak bases, therefore, do not directly inhibit ASM, but result in a functional inhibition of ASM. We have thus proposed the acronym FIASMA for Functional Inhibitor of Acid SphingoMyelinAse. According to this model, functional inhibition of ASM requires high lysosomal concentrations of a weak basic drug. Previously, we have shown that functional inhibition of ASM is related to high pKa- and high logP-values and have characterized several new FIASMAs, including the antidepressant drugs doxepine 63, fluoxetine 104, maprotilin 109, nortriptyline 114, paroxetine 118 and sertraline 124. The aims of the present study wereto identify more FIASMAs,to further improve the in silico prediction of functional ASM inhibition by developing compact and easily-interpretable models with high internal 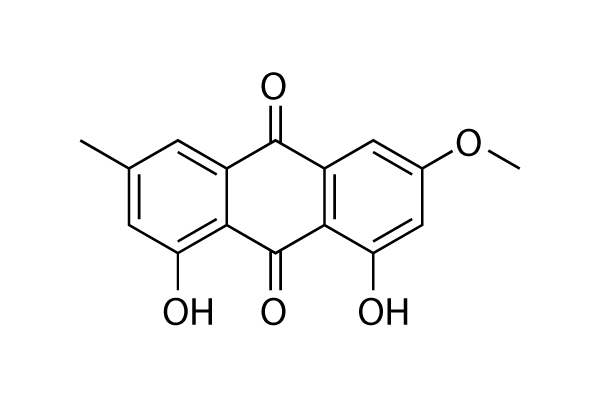 CDE. The expression of mutants of proteins involved in the clathrin machinery, such as Dynamin2-K44A, the carboxy terminus of AP180, and clathrin hubs, has proven quite effective. More recently siRNA-mediated depletion of the clathrin heavy chain, subunits of the AP2 adaptor, and dynamin 2have abolished CDE in cells. The drawback of these genetic approaches is that they require days to take effect and may lead to many indirect effects or compensatory cellular responses that make interpretation of the findings sometimes difficult. Use of a number of acute cellular treatments including cytosol acidification and hypotonic treatment can be effective at blocking endocytosis of CDE cargobut these treatments are non-specific and may also affect CIE. Recently, new compounds that selectively target proteins involved in CDE have been identified with the promise that these could be used to acutely inhibit this process. These include compounds that specifically target dynamin such as dynasoreand the dynoles. Since dynamin is required for all forms of CDE and is used in some forms of CIE, a compound that selectively targets clathrin was developed by Haucke and colleagues. This compound, named pitstop 2, was designed and shown to bind to and block interactions between the amino terminal domain of clathrin heavy chain and amphiphysin, one of many proteins shown to bind to this domain of clathrin. In cells, pitstop 2 was shown to inhibit endocytosis of transferrin receptor, a CDE cargo
CDE. The expression of mutants of proteins involved in the clathrin machinery, such as Dynamin2-K44A, the carboxy terminus of AP180, and clathrin hubs, has proven quite effective. More recently siRNA-mediated depletion of the clathrin heavy chain, subunits of the AP2 adaptor, and dynamin 2have abolished CDE in cells. The drawback of these genetic approaches is that they require days to take effect and may lead to many indirect effects or compensatory cellular responses that make interpretation of the findings sometimes difficult. Use of a number of acute cellular treatments including cytosol acidification and hypotonic treatment can be effective at blocking endocytosis of CDE cargobut these treatments are non-specific and may also affect CIE. Recently, new compounds that selectively target proteins involved in CDE have been identified with the promise that these could be used to acutely inhibit this process. These include compounds that specifically target dynamin such as dynasoreand the dynoles. Since dynamin is required for all forms of CDE and is used in some forms of CIE, a compound that selectively targets clathrin was developed by Haucke and colleagues. This compound, named pitstop 2, was designed and shown to bind to and block interactions between the amino terminal domain of clathrin heavy chain and amphiphysin, one of many proteins shown to bind to this domain of clathrin. In cells, pitstop 2 was shown to inhibit endocytosis of transferrin receptor, a CDE cargo  inhibitor when used at high doses. Phosphorylation of these kinases has been reported to be regulated negatively by O-GlcNAcylation. It is worth noting that different effects of OGA inhibition on phosphorylation of AKT and GSK-3 have been reported. Elevation of O-GlcNAcylation in skeletal muscles after OGA inhibition using another inhibitor, PUGNAc, does not significantly alter insulin-stimulated phosphorylation of AKT or GSK-3. In differentiated 3T3-L1 adipocytes, two different OGA inhibitors have been found to increase O-GlcNAc levels but not alter insulin-stimulated phosphorylation of AKT nor induce insulin resistance either. Therefore, it remains somewhat unclear as to the effects of OGA inhibition on alteration of GSK3b levels and AKT activity; the effects observed here could stem from high-dose inhibition of OGA, or alternatively from off-target effects of using the inhibitor at a high dose. Tau is abnormally hyperphosphorylated and aggregated in AD and other tauopathies. Previous studies from our and other groups have demonstrated differential roles of tau phosphorylation at various phosphorylation sites. A quantitative in vitro study demonstrated that phosphorylation of tau at Ser262, Thr231, and Ser235 inhibits its binding to microtubules by,35%,,25%, and,10%, respectively. In vitro kinetic studies of the binding between hyperphosphorylated tau and normal tau suggest that Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/ Ser356 and Ser422 are among the critical phosphorylation sites that convert tau to an inhibitory molecule that sequesters normal microtubule-associated proteins from microtubules. Further phosphorylation at Thr231, Ser396, and Ser422 promotes selfaggregation of tau into filaments. It is obvious that tau phosphorylation at various sites impacts tau activity and aggregation collectively. Our recent study has demonstrated that tau phosphorylation at the proline-rich region, which is located upstream of the microtubule-binding domains, inhibits its microtubule
inhibitor when used at high doses. Phosphorylation of these kinases has been reported to be regulated negatively by O-GlcNAcylation. It is worth noting that different effects of OGA inhibition on phosphorylation of AKT and GSK-3 have been reported. Elevation of O-GlcNAcylation in skeletal muscles after OGA inhibition using another inhibitor, PUGNAc, does not significantly alter insulin-stimulated phosphorylation of AKT or GSK-3. In differentiated 3T3-L1 adipocytes, two different OGA inhibitors have been found to increase O-GlcNAc levels but not alter insulin-stimulated phosphorylation of AKT nor induce insulin resistance either. Therefore, it remains somewhat unclear as to the effects of OGA inhibition on alteration of GSK3b levels and AKT activity; the effects observed here could stem from high-dose inhibition of OGA, or alternatively from off-target effects of using the inhibitor at a high dose. Tau is abnormally hyperphosphorylated and aggregated in AD and other tauopathies. Previous studies from our and other groups have demonstrated differential roles of tau phosphorylation at various phosphorylation sites. A quantitative in vitro study demonstrated that phosphorylation of tau at Ser262, Thr231, and Ser235 inhibits its binding to microtubules by,35%,,25%, and,10%, respectively. In vitro kinetic studies of the binding between hyperphosphorylated tau and normal tau suggest that Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/ Ser356 and Ser422 are among the critical phosphorylation sites that convert tau to an inhibitory molecule that sequesters normal microtubule-associated proteins from microtubules. Further phosphorylation at Thr231, Ser396, and Ser422 promotes selfaggregation of tau into filaments. It is obvious that tau phosphorylation at various sites impacts tau activity and aggregation collectively. Our recent study has demonstrated that tau phosphorylation at the proline-rich region, which is located upstream of the microtubule-binding domains, inhibits its microtubule 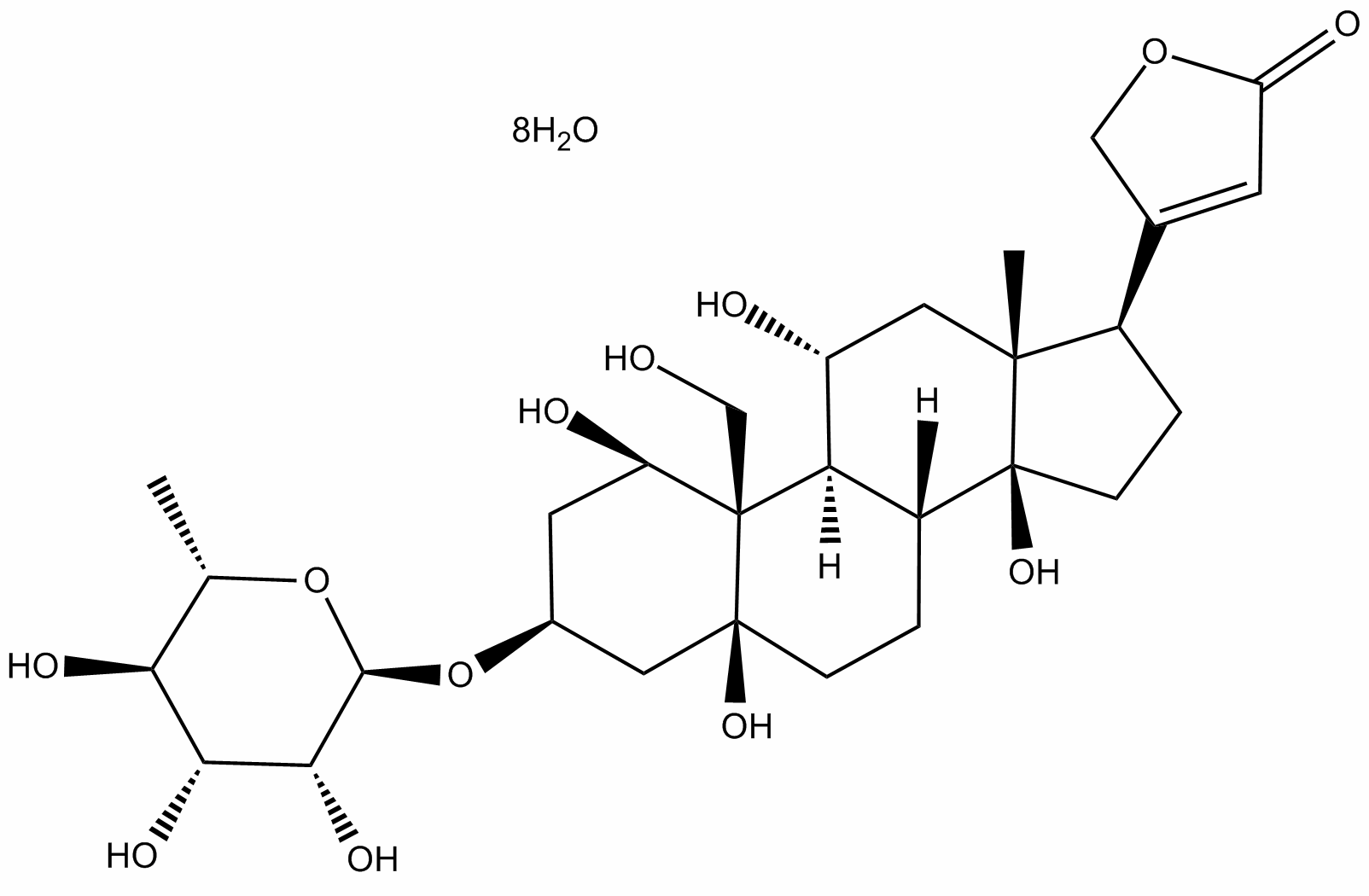 is regulated by PI3K and other factors. We found that thiamet-G treatment did not significantly alter the level of GSK-3b except after treatment for 9�C24 h, but blocked its phosphorylation at Ser9 completely, suggesting that GSK-3b was markedly activated under these conditions. Phosphorylation of GSK-3b at Tyr216, which makes it more active, was also increased 24 h after thiamet-G treatment. Consistent with the almost complete dephosphorylation of GSK-3b at Ser9, Ser473 and Thr308 phosphorylation of AKT, which determines its kinase activity, was also blocked by thiamet-G treatment. However, we did not find any significant changes of either the level or the activation of PI3K, the major upstream kinase of AKT, in the mouse brain after thiamet-G treatment. These results suggest that thiamet-G induced over-activation of GSK-3b via inhibition of AKT phosphorylation. The over-activation of GSK-3b may explain the increased tau phosphorylation at Ser199, Ser202, Ser396 and Ser422,
is regulated by PI3K and other factors. We found that thiamet-G treatment did not significantly alter the level of GSK-3b except after treatment for 9�C24 h, but blocked its phosphorylation at Ser9 completely, suggesting that GSK-3b was markedly activated under these conditions. Phosphorylation of GSK-3b at Tyr216, which makes it more active, was also increased 24 h after thiamet-G treatment. Consistent with the almost complete dephosphorylation of GSK-3b at Ser9, Ser473 and Thr308 phosphorylation of AKT, which determines its kinase activity, was also blocked by thiamet-G treatment. However, we did not find any significant changes of either the level or the activation of PI3K, the major upstream kinase of AKT, in the mouse brain after thiamet-G treatment. These results suggest that thiamet-G induced over-activation of GSK-3b via inhibition of AKT phosphorylation. The over-activation of GSK-3b may explain the increased tau phosphorylation at Ser199, Ser202, Ser396 and Ser422,