The molecular events implicated in repair of CT99021 252917-06-9 strand breaks in DNA are becoming more clear, but an overall and quantitative picture of their repair in vivo which would contribute to understanding the systems biology of repair and the effects of inhibitors is not yet available. Current methods do not allow simultaneous and precise quantitation of repair of Perifosine single and double strand breaks. Repair of double strand breaks, which are believed to be the crucial lesions leading to cell death, is commonly assayed by restoration of the normal length of genomic DNA or restriction fragments using pulsed-field gel electrophoresis. Repair of single strand breaks, which may contribute to loss of viability by relaxing superhelical stress in genomic DNA loops and thus arresting transcription, cannot yet be quantitated specifically by methods with comparable precision. As a model system to approach this question we are studying the repair of strand breaks in vivo in a,170 kb circular minichromosome, the Epstein-Barr virus episome, which is maintained in the nuclei of Raji cells at 50�C100 copies localised at the periphery of interphase chromosomes. Two features of this minichromosome make it an attractive model for genomic chromatin: it can be considered as a defined region of chromatin in view of its canonical nucleosomal conformation and the well-studied sequence and properties of its DNA, and its closed circular topology and length resemble those of the constrained loops which genomic chromatin forms in vivo. After irradiating cells with 60Co c photons we assayed the repair of single strand breaks in the minichromosome by quantitating the loss of nuclease S1sensitive sites, and the repair of double strand breaks by PFGE assays of the reformation of supercoiled DNA from molecules which had been linearised. Circular molecules containing single strand breaks could not be quantitated directly, and instead their levels were calculated using a mathematical model developed to fit the experimental data. We exploited the possibility of quantitating repair in this system to examine the implication of particular enzymes, particularly topoisomerases I and II whose participation in repair has long been controversial, poly polymerase-1, Rad51, the catalytic subunit of DNA-protein kinase, and ATM kinase. New features of the repair of strand breaks in vivo and of their kinetics were revealed by mathematical modeling. The simultaneous repair of single and double strand breaks in a defined region of chromatin in vivo has not been studied previously using quantitative methods, to our knowledge. The methods used to detect strand breaks in earlier studies, filter elution or single-cell DNA electrophoresis, cannot provide absolute numbers of breaks and the reported rates were variable. We used two conditions to ensure that strand breaks were quantitated accurately: for PFGE, DNA was deproteinised at room temperature because extra strand breaks are created at higher temperatures, and hybridisation was carried out  in dried gels because the transfer of large DNA fragments onto membranes is not quantitative. In another study published while this manuscript was in preparation, a significant amount of minichromosome DNA remained in the sample well of PFGE gels and was interpreted as nicked circles, but here little or no DNA remained in the wells and nicked circular DNA migrated slowly into the gel, possibly reflecting methodological differences.
in dried gels because the transfer of large DNA fragments onto membranes is not quantitative. In another study published while this manuscript was in preparation, a significant amount of minichromosome DNA remained in the sample well of PFGE gels and was interpreted as nicked circles, but here little or no DNA remained in the wells and nicked circular DNA migrated slowly into the gel, possibly reflecting methodological differences.
A Poisson distribution of strand breaks was assumed DPP-IV inihbitors alone can reverse new-onset diabetes
Leave a reply
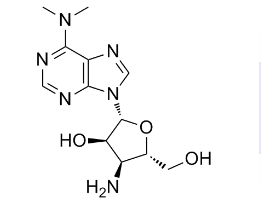 by ligases IIIb and IV-XRCC4 which participate in NHEJ. Kinetic models of strand break repair can be constructed with different degrees of complexity, but theory shows that the least complex model is preferable to provide concrete predictions. Our data were fitted well by using first-order kinetics, and we consider that this strategy was justified since other datasets for DNA repair have been fitted satisfactorily by first-order kinetics, which only deviate significantly from higherorder models after two half-times ; further, theoretical arguments show that “multiple processes may combine to produce kinetic behavior indistinguishable from firstorder and. are more likely to exist when reactions occur in a complex environment”. A number of conclusions which were not directly apparent from the experimental data
by ligases IIIb and IV-XRCC4 which participate in NHEJ. Kinetic models of strand break repair can be constructed with different degrees of complexity, but theory shows that the least complex model is preferable to provide concrete predictions. Our data were fitted well by using first-order kinetics, and we consider that this strategy was justified since other datasets for DNA repair have been fitted satisfactorily by first-order kinetics, which only deviate significantly from higherorder models after two half-times ; further, theoretical arguments show that “multiple processes may combine to produce kinetic behavior indistinguishable from firstorder and. are more likely to exist when reactions occur in a complex environment”. A number of conclusions which were not directly apparent from the experimental data 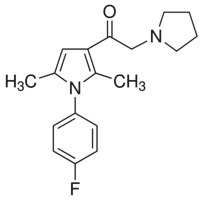 is thought to be a key contributor in the development of castration resistance, as well as general resistance to anti-cancer treatments. Elucidating the role of anti-apoptotic genes/proteins in the progression of prostate cancer is therefore likely to lead to improvements in the treatment of refractory disease. The Inhibitors of Apoptosis Protein family has been reported to play a role in apoptosis resistance in a variety of cancer cell lines and is characterized by the presence in the proteins of one to three copies of a Baculoviral IAP Repeat domain. The IAPs have been demonstrated to bind to and inhibit a variety of pro-apoptotic factors, thereby effectively suppressing apoptosis induced by a wide range of effectors, including chemotherapeutics and irradiation. The BIR domain is essential for interaction of the IAPs with pro-apoptotic factors, including caspases. The caspases are a family of cysteineaspartic acid-specific proteases, present in a pro-form which, once activated via cleavage, is responsible for degradation of death substrates such as poly-ADP-ribose polymerase thereby triggering apoptosis. Cleaved caspase-3 and cleaved PARP can be readily detected by Western blot analysis and are commonly used as markers for apoptosis. Apoptosis is often associated with autophagy, a process involving lysosomal degradation of a cell��s own components. It involves packaging of proteins and organelles within autophagosomes, followed by fusion with lysosomes leading to degradation of the proteins and organelles. The role of autophagy in the development of cancer and its treatment is complex, since there is evidence that autophagy can promote and suppress cancer growth. Inhibition of autophagy by disruption of essential autophagy genes has been shown to promote tumorigenesis and hence autophagy can have a tumor-suppressive effect. However, there is increasing evidence that autophagy can act as a survival mechanism for cancer cells in response to a wide range of stresses, including treatment with anti-cancer agents. To detect autophagic activity in cultured cells, Western blot detection of LC3B-II is often used. LC3B-II is specifically associated with autophagosomes and levels of LC3B-II have been demonstrated to correlate with the number of autophagosomes within cells. However, since LC3B-II is degraded upon autophagosome-lysosome fusion.
is thought to be a key contributor in the development of castration resistance, as well as general resistance to anti-cancer treatments. Elucidating the role of anti-apoptotic genes/proteins in the progression of prostate cancer is therefore likely to lead to improvements in the treatment of refractory disease. The Inhibitors of Apoptosis Protein family has been reported to play a role in apoptosis resistance in a variety of cancer cell lines and is characterized by the presence in the proteins of one to three copies of a Baculoviral IAP Repeat domain. The IAPs have been demonstrated to bind to and inhibit a variety of pro-apoptotic factors, thereby effectively suppressing apoptosis induced by a wide range of effectors, including chemotherapeutics and irradiation. The BIR domain is essential for interaction of the IAPs with pro-apoptotic factors, including caspases. The caspases are a family of cysteineaspartic acid-specific proteases, present in a pro-form which, once activated via cleavage, is responsible for degradation of death substrates such as poly-ADP-ribose polymerase thereby triggering apoptosis. Cleaved caspase-3 and cleaved PARP can be readily detected by Western blot analysis and are commonly used as markers for apoptosis. Apoptosis is often associated with autophagy, a process involving lysosomal degradation of a cell��s own components. It involves packaging of proteins and organelles within autophagosomes, followed by fusion with lysosomes leading to degradation of the proteins and organelles. The role of autophagy in the development of cancer and its treatment is complex, since there is evidence that autophagy can promote and suppress cancer growth. Inhibition of autophagy by disruption of essential autophagy genes has been shown to promote tumorigenesis and hence autophagy can have a tumor-suppressive effect. However, there is increasing evidence that autophagy can act as a survival mechanism for cancer cells in response to a wide range of stresses, including treatment with anti-cancer agents. To detect autophagic activity in cultured cells, Western blot detection of LC3B-II is often used. LC3B-II is specifically associated with autophagosomes and levels of LC3B-II have been demonstrated to correlate with the number of autophagosomes within cells. However, since LC3B-II is degraded upon autophagosome-lysosome fusion.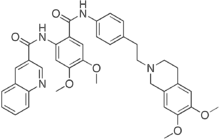 that the reduction in cell viability was attributable to apoptosis. In this study, a decrease in cell viability induced by BIRC6 reduction did not confine to cells expressing wild-type p53, contrary to previous reports suggesting that apoptosis resulting from BIRC6 knockdown in H460 cells and breast cancer cells requires functional p53. We showed that both wild type p53 and p53 null cells were sensitive to BIRC6 siRNA induced growth inhibition. This variation reflects that apoptosis induction by loss of BIRC6 may be facilitated by different mechanisms in different models. Further investigation is necessary to understand the underlying mechanism leading to apoptosis after BIRC6 reduction in p53 null cells and that will provide further insight in the possibility of targeting BIRC6 in cancer cells lacking functional p53. Elevated levels of BIRC6 have been linked to apoptosis resistance, for instance in the SNB-78
that the reduction in cell viability was attributable to apoptosis. In this study, a decrease in cell viability induced by BIRC6 reduction did not confine to cells expressing wild-type p53, contrary to previous reports suggesting that apoptosis resulting from BIRC6 knockdown in H460 cells and breast cancer cells requires functional p53. We showed that both wild type p53 and p53 null cells were sensitive to BIRC6 siRNA induced growth inhibition. This variation reflects that apoptosis induction by loss of BIRC6 may be facilitated by different mechanisms in different models. Further investigation is necessary to understand the underlying mechanism leading to apoptosis after BIRC6 reduction in p53 null cells and that will provide further insight in the possibility of targeting BIRC6 in cancer cells lacking functional p53. Elevated levels of BIRC6 have been linked to apoptosis resistance, for instance in the SNB-78 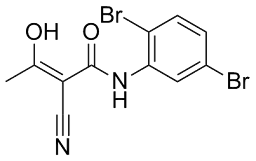 1. This table indicates that IC50 values vary from 0.5 nM to 36 nM and SIE values calculated from this work are in the range 210.03 kcal/mol to 210.67 kcal/mol. Though there is no direct correlation between IC50 and SIE values, it can be observed that their respective values lie within a narrow range. Many patients eventually developed imatinib resistance, usually associated with above mentioned mutations in ABL kinase domain that either directly or indirectly effects the binding affinity of imatinib to ABL. The most common gatekeeper residue mutation T315I that accounts for 15�C20% of clinically observed mutations is completely resistant to imatinib, nilotinib and dasatinib. Native and T315I BCR-ABL kinases complexed with dasatinib are subjected to 25 ns of MD simulations and SIE binding free energies are calculated. The analysis of dasatinib complexed with native and T315I mutant BCR-ABL kinases revealed that native complex has relatively higher SIE free energy than when complexed with T315I that signifies the greater affinity of dasatinib for native compared to mutant BCR-ABL kinase.
1. This table indicates that IC50 values vary from 0.5 nM to 36 nM and SIE values calculated from this work are in the range 210.03 kcal/mol to 210.67 kcal/mol. Though there is no direct correlation between IC50 and SIE values, it can be observed that their respective values lie within a narrow range. Many patients eventually developed imatinib resistance, usually associated with above mentioned mutations in ABL kinase domain that either directly or indirectly effects the binding affinity of imatinib to ABL. The most common gatekeeper residue mutation T315I that accounts for 15�C20% of clinically observed mutations is completely resistant to imatinib, nilotinib and dasatinib. Native and T315I BCR-ABL kinases complexed with dasatinib are subjected to 25 ns of MD simulations and SIE binding free energies are calculated. The analysis of dasatinib complexed with native and T315I mutant BCR-ABL kinases revealed that native complex has relatively higher SIE free energy than when complexed with T315I that signifies the greater affinity of dasatinib for native compared to mutant BCR-ABL kinase.