In genomic DNA the fraction of double strand breaks repaired by HR varies in different cell types and is predominant in lower eukaryotes, whose smaller genome may allow homologous chromosomes to find each other more easily than those in higher eukaryotes. Similarly, HR may be favoured in the minichromosome due to the proximity of numerous replicating and daughter DNA molecules in replication compartments whose limited volume would facilitate finding a region of sequence homology in a neighbouring molecule. Linear oligomers of minichromosome DNA were not detected during repair, as also observed during repair of a 3 Mb double-minute chromosome and transfected plasmids, reflecting juxtaposition of the extremities of the broken DNA by Ku and the RMX complex; we propose that a further important factor is the crowded macromolecular Torin 1 environment in the nucleus because crowding strongly favours DNA circularisation and ligation 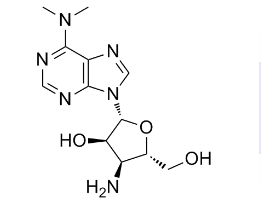 by ligases IIIb and IV-XRCC4 which participate in NHEJ. Kinetic models of strand break repair can be constructed with different degrees of complexity, but theory shows that the least complex model is preferable to provide concrete predictions. Our data were fitted well by using first-order kinetics, and we consider that this strategy was justified since other datasets for DNA repair have been fitted satisfactorily by first-order kinetics, which only deviate significantly from higherorder models after two half-times ; further, theoretical arguments show that “multiple processes may combine to produce kinetic behavior indistinguishable from firstorder and. are more likely to exist when reactions occur in a complex environment”. A number of conclusions which were not directly apparent from the experimental data LY2157299 illustrated the usefullness of modeling. First, when repair of double strand breaks was arrested, the single strand breaks in linear molecules were still repaired and circular molecules containing single strand breaks were converted to supercoiled molecules at close to the normal rate showing that the systems which repair single and double strand breaks operate independently, which has not been demonstrated previously as far as we are aware. Second, the calculated rate constants show that in an average linearised minichromosome the double strand break was repaired three to four times faster than all the single strand breaks, so that the ratelimiting step for complete repair of minichromosomes was the repair of single strand breaks. These repair rates cannot be compaired directly with those reported for genomic DNA where the methods used could not quantitate breaks directly, but comparisons can be made in terms of the half-time for repair which is independent of the radiation dose and of the length of the region considered. In the minichromosome the calculated half-time for repair of the double strand break in each molecule was,40 min, which is within the range of 20 to 110 min reported for genomic DNA. For repair of single strand breaks the half-time of,140 min for repairing 8 to 9 breaks per molecule was equivalent to an average of,16 min/break, which is within the range reported for genomic DNA. This minichromosome offers an simple experimental system for quantitative testing of potential inhibitors of repair of strand breaks, and since the sequence and structural features of its DNA and its transcription pattern have been studied extensively.
by ligases IIIb and IV-XRCC4 which participate in NHEJ. Kinetic models of strand break repair can be constructed with different degrees of complexity, but theory shows that the least complex model is preferable to provide concrete predictions. Our data were fitted well by using first-order kinetics, and we consider that this strategy was justified since other datasets for DNA repair have been fitted satisfactorily by first-order kinetics, which only deviate significantly from higherorder models after two half-times ; further, theoretical arguments show that “multiple processes may combine to produce kinetic behavior indistinguishable from firstorder and. are more likely to exist when reactions occur in a complex environment”. A number of conclusions which were not directly apparent from the experimental data LY2157299 illustrated the usefullness of modeling. First, when repair of double strand breaks was arrested, the single strand breaks in linear molecules were still repaired and circular molecules containing single strand breaks were converted to supercoiled molecules at close to the normal rate showing that the systems which repair single and double strand breaks operate independently, which has not been demonstrated previously as far as we are aware. Second, the calculated rate constants show that in an average linearised minichromosome the double strand break was repaired three to four times faster than all the single strand breaks, so that the ratelimiting step for complete repair of minichromosomes was the repair of single strand breaks. These repair rates cannot be compaired directly with those reported for genomic DNA where the methods used could not quantitate breaks directly, but comparisons can be made in terms of the half-time for repair which is independent of the radiation dose and of the length of the region considered. In the minichromosome the calculated half-time for repair of the double strand break in each molecule was,40 min, which is within the range of 20 to 110 min reported for genomic DNA. For repair of single strand breaks the half-time of,140 min for repairing 8 to 9 breaks per molecule was equivalent to an average of,16 min/break, which is within the range reported for genomic DNA. This minichromosome offers an simple experimental system for quantitative testing of potential inhibitors of repair of strand breaks, and since the sequence and structural features of its DNA and its transcription pattern have been studied extensively.
Particular pathway of double strand break repair which is arrested when DNA-PKcs is inhibited
Leave a reply
 is thought to be a key contributor in the development of castration resistance, as well as general resistance to anti-cancer treatments. Elucidating the role of anti-apoptotic genes/proteins in the progression of prostate cancer is therefore likely to lead to improvements in the treatment of refractory disease. The Inhibitors of Apoptosis Protein family has been reported to play a role in apoptosis resistance in a variety of cancer cell lines and is characterized by the presence in the proteins of one to three copies of a Baculoviral IAP Repeat domain. The IAPs have been demonstrated to bind to and inhibit a variety of pro-apoptotic factors, thereby effectively suppressing apoptosis induced by a wide range of effectors, including chemotherapeutics and irradiation. The BIR domain is essential for interaction of the IAPs with pro-apoptotic factors, including caspases. The caspases are a family of cysteineaspartic acid-specific proteases, present in a pro-form which, once activated via cleavage, is responsible for degradation of death substrates such as poly-ADP-ribose polymerase thereby triggering apoptosis. Cleaved caspase-3 and cleaved PARP can be readily detected by Western blot analysis and are commonly used as markers for apoptosis. Apoptosis is often associated with autophagy, a process involving lysosomal degradation of a cell��s own components. It involves packaging of proteins and organelles within autophagosomes, followed by fusion with lysosomes leading to degradation of the proteins and organelles. The role of autophagy in the development of cancer and its treatment is complex, since there is evidence that autophagy can promote and suppress cancer growth. Inhibition of autophagy by disruption of essential autophagy genes has been shown to promote tumorigenesis and hence autophagy can have a tumor-suppressive effect. However, there is increasing evidence that autophagy can act as a survival mechanism for cancer cells in response to a wide range of stresses, including treatment with anti-cancer agents. To detect autophagic activity in cultured cells, Western blot detection of LC3B-II is often used. LC3B-II is specifically associated with autophagosomes and levels of LC3B-II have been demonstrated to correlate with the number of autophagosomes within cells. However, since LC3B-II is degraded upon autophagosome-lysosome fusion.
is thought to be a key contributor in the development of castration resistance, as well as general resistance to anti-cancer treatments. Elucidating the role of anti-apoptotic genes/proteins in the progression of prostate cancer is therefore likely to lead to improvements in the treatment of refractory disease. The Inhibitors of Apoptosis Protein family has been reported to play a role in apoptosis resistance in a variety of cancer cell lines and is characterized by the presence in the proteins of one to three copies of a Baculoviral IAP Repeat domain. The IAPs have been demonstrated to bind to and inhibit a variety of pro-apoptotic factors, thereby effectively suppressing apoptosis induced by a wide range of effectors, including chemotherapeutics and irradiation. The BIR domain is essential for interaction of the IAPs with pro-apoptotic factors, including caspases. The caspases are a family of cysteineaspartic acid-specific proteases, present in a pro-form which, once activated via cleavage, is responsible for degradation of death substrates such as poly-ADP-ribose polymerase thereby triggering apoptosis. Cleaved caspase-3 and cleaved PARP can be readily detected by Western blot analysis and are commonly used as markers for apoptosis. Apoptosis is often associated with autophagy, a process involving lysosomal degradation of a cell��s own components. It involves packaging of proteins and organelles within autophagosomes, followed by fusion with lysosomes leading to degradation of the proteins and organelles. The role of autophagy in the development of cancer and its treatment is complex, since there is evidence that autophagy can promote and suppress cancer growth. Inhibition of autophagy by disruption of essential autophagy genes has been shown to promote tumorigenesis and hence autophagy can have a tumor-suppressive effect. However, there is increasing evidence that autophagy can act as a survival mechanism for cancer cells in response to a wide range of stresses, including treatment with anti-cancer agents. To detect autophagic activity in cultured cells, Western blot detection of LC3B-II is often used. LC3B-II is specifically associated with autophagosomes and levels of LC3B-II have been demonstrated to correlate with the number of autophagosomes within cells. However, since LC3B-II is degraded upon autophagosome-lysosome fusion. that the reduction in cell viability was attributable to apoptosis. In this study, a decrease in cell viability induced by BIRC6 reduction did not confine to cells expressing wild-type p53, contrary to previous reports suggesting that apoptosis resulting from BIRC6 knockdown in H460 cells and breast cancer cells requires functional p53. We showed that both wild type p53 and p53 null cells were sensitive to BIRC6 siRNA induced growth inhibition. This variation reflects that apoptosis induction by loss of BIRC6 may be facilitated by different mechanisms in different models. Further investigation is necessary to understand the underlying mechanism leading to apoptosis after BIRC6 reduction in p53 null cells and that will provide further insight in the possibility of targeting BIRC6 in cancer cells lacking functional p53. Elevated levels of BIRC6 have been linked to apoptosis resistance, for instance in the SNB-78
that the reduction in cell viability was attributable to apoptosis. In this study, a decrease in cell viability induced by BIRC6 reduction did not confine to cells expressing wild-type p53, contrary to previous reports suggesting that apoptosis resulting from BIRC6 knockdown in H460 cells and breast cancer cells requires functional p53. We showed that both wild type p53 and p53 null cells were sensitive to BIRC6 siRNA induced growth inhibition. This variation reflects that apoptosis induction by loss of BIRC6 may be facilitated by different mechanisms in different models. Further investigation is necessary to understand the underlying mechanism leading to apoptosis after BIRC6 reduction in p53 null cells and that will provide further insight in the possibility of targeting BIRC6 in cancer cells lacking functional p53. Elevated levels of BIRC6 have been linked to apoptosis resistance, for instance in the SNB-78