SFA monotherapy did not suppress acute organ allograft rejection supporting the hypothesis that it does not represent a primary T cell inhibitor. Interestingly, in combination with CsA, SFA efficiently promoted long-term allograft survival. Furthermore, in a chronic allograft rejection model, addition of SFA to CsA-treated recipients markedly inhibited chronic rejection compared to animals receiving high dose CsA monotherapy, suggesting that SFA exerted unique immunobiological effects different from DAPT inhibition of calcineurin phosphatases. Current knowledge indicates that SFA represents a novel class of immunophilin-binding metabolite both with respect to chemical structure and functional activity. There is a paucity of knowledge about the immunobiological effects of SFA since each study focused on selected functions or selected aspects with professional antigen presenting cells being either directly or indirectly involved. Systematic studies investigating the effects of SFA are completely lacking. In this report we describe the results of the first systematic analysis of the immunobiological effects of the novel immunophilin-binding agent SFA on human monocyte-derived DC using a combination of genome-wide expression profiling with subsequent confirmation on the protein level and functional in vitro and in vivo assays. Results indicate that SFA represents a novel DC CX-4945 1009820-21-6 chemokine and migration inhibitor. Sanglifehrins represent novel immunosuppressive agents that have been reported to suppress key functions of DCs. We and others have reported that SFA inhibits bioactive IL-12p70 production, macropinocytosis as well as receptor-mediated endocytosis in human and murine DCs. Transplant experiments indicated that addition of SFA to CsA efficiently suppresses graft arteriosclerosis in comparison to CsA monotherapy suggesting that SFA may represent a novel class of immunophilin-binding agents. However, a disadvantage of previous studies is the fact that they have focused on selected molecules or selected functional aspects thereby restricting the possibility to discover novel mechanisms of action. Accordingly, the aim of the present study was to use a systematic genome-wide approach in order to reveal novel immunobiological effects of SFA on human DC. Secondly, identification of molecules being most specifically suppressed by SFA in comparison to the related molecule CsA may help to elucidate the mechanism of action. The results presented here indicate that SFA impairs DCmediated immunity in a so far unrecognized manner that is DC chemokine expression and migration. Importantly, SFA��s inhibitory effects can be demonstrated on two different functional levels such as direct chemokine expression inhibition and subsequent impaired attraction of CD4 helper T cells as wells as DC migration inhibition towards recombinant CCL19. Accordingly, we have found that SFA, in contrast to CsA, does not only inhibit mRNA and protein expression of a number of chemokines, including CCL5, CCL17 and CCL19 but additionally suppresses CD38 mRNA and DC surface expression. Thus, SFA��s effects on DC are unique in direct comparison to the related cyclophilin-binding immunosuppressant CsA. The latter results provide a rationale for the explanation of reduced migration of SFA-exposed moDCs against recombinant CCL19. CD38 has been reported to be required forthe migration of mature DC against recombinant CCL19. Furthermore, CD38 inhibition by SFA provides additional insight into recent reports demonstrating SFA��s capacity to abrogate bioactive IL-12 production in vitro and 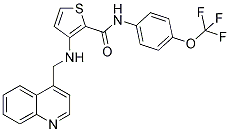 in vivo.
in vivo.
involved in IL-12 production and IL-12 secretion has been demonstrated to be restored upon CD38
Leave a reply
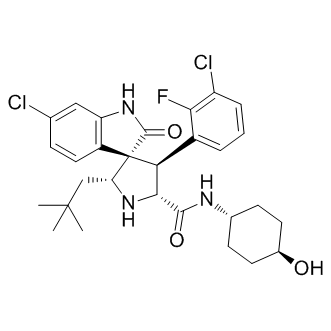 could act elsewhere and ultimately affect the respiratory parameters of the mitochondria. Perhaps the broadest consideration that must be taken into account is the possibility that most if not all the HDACis effects described in the present work could have been brought about by an action on a central factor that, in turn, would affect other pathways. One possibility is that HDACis are decreasing HIF-1a, and this effect decreases glycolysis maintained by HIF-1a. A key transcription factor that knowingly acts as the hub for many processes associated to the cell cycle and the energy metabolism itself. Thus, by preferentially targeting HIF-1a, NaB or TSA could indirectly affect hundreds of other genes. At any rate, investigation of the effect of HDACis on HIF1a regulation and on key actors of anaerobic and oxidative metabolism is currently under way. Although more evidence has yet to be procured, the set of results contained herein suggests that NaB and TSA could induce H460 cells to assume an altogether differentiated state. It is proposed that in this differentiated phenotype, cells actually shift towards more oxidative metabolism akin to untransformed cells. Ultimately, the enhanced oxidative metabolism would harm the H460 cells by way of excessive production of ROS that can cause cell death. In this context, the H460 cells treated with NaB could activate antioxidant defense mechanisms related to the increase in: HK activity bound to mitochondria, which increases ADP recycling; G6PDH activity, which increases PPP flux and NADPH supply; and the expression of Mfn 1, which, may reflect the initial stages of a process of mitochondrial fusion that can ultimately act as inhibitor of apoptosis. Indeed it has been shown that under oxidative stress mitochondria can rescue their membrane potential. Although the notion that NaB and TSA induced generation of ROS is a reasonable proposition, it must be mentioned that the topic of ROS and cancer cells is in itself quite controversial. There are data supporting the view that ROS are in fact essential requirements for the survival of tumor cells, even though the mechanism whereby ROS would support tumor growth remains elusive. Taken together the results presented here revealed a unique biochemical profile induced by NaB and TSA. The data essentially showed that alterations in the glycolytic flux triggered an enhancement of mitochondrial function that was not, however, paralleled by cell proliferation. Whilst further studies are paramount to unveil the mechanisms underlying the interplay between glycolysis and oxidative metabolism, the groundwork established in the present work already suggests that the stimulation of the oxidative metabolism in tumor cells may be an interesting strategy for chemotherapy. Ligand-based virtual screening, quantitative structureproperty and structure-activity relationships, and other concepts in computational medicinal chemistry are based on the similarity principle, which states that similar compounds generally exhibit similar properties.
could act elsewhere and ultimately affect the respiratory parameters of the mitochondria. Perhaps the broadest consideration that must be taken into account is the possibility that most if not all the HDACis effects described in the present work could have been brought about by an action on a central factor that, in turn, would affect other pathways. One possibility is that HDACis are decreasing HIF-1a, and this effect decreases glycolysis maintained by HIF-1a. A key transcription factor that knowingly acts as the hub for many processes associated to the cell cycle and the energy metabolism itself. Thus, by preferentially targeting HIF-1a, NaB or TSA could indirectly affect hundreds of other genes. At any rate, investigation of the effect of HDACis on HIF1a regulation and on key actors of anaerobic and oxidative metabolism is currently under way. Although more evidence has yet to be procured, the set of results contained herein suggests that NaB and TSA could induce H460 cells to assume an altogether differentiated state. It is proposed that in this differentiated phenotype, cells actually shift towards more oxidative metabolism akin to untransformed cells. Ultimately, the enhanced oxidative metabolism would harm the H460 cells by way of excessive production of ROS that can cause cell death. In this context, the H460 cells treated with NaB could activate antioxidant defense mechanisms related to the increase in: HK activity bound to mitochondria, which increases ADP recycling; G6PDH activity, which increases PPP flux and NADPH supply; and the expression of Mfn 1, which, may reflect the initial stages of a process of mitochondrial fusion that can ultimately act as inhibitor of apoptosis. Indeed it has been shown that under oxidative stress mitochondria can rescue their membrane potential. Although the notion that NaB and TSA induced generation of ROS is a reasonable proposition, it must be mentioned that the topic of ROS and cancer cells is in itself quite controversial. There are data supporting the view that ROS are in fact essential requirements for the survival of tumor cells, even though the mechanism whereby ROS would support tumor growth remains elusive. Taken together the results presented here revealed a unique biochemical profile induced by NaB and TSA. The data essentially showed that alterations in the glycolytic flux triggered an enhancement of mitochondrial function that was not, however, paralleled by cell proliferation. Whilst further studies are paramount to unveil the mechanisms underlying the interplay between glycolysis and oxidative metabolism, the groundwork established in the present work already suggests that the stimulation of the oxidative metabolism in tumor cells may be an interesting strategy for chemotherapy. Ligand-based virtual screening, quantitative structureproperty and structure-activity relationships, and other concepts in computational medicinal chemistry are based on the similarity principle, which states that similar compounds generally exhibit similar properties.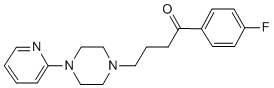 results highlight the cross-reactivity of several kinase inhibitors with DDK and also reveal an opportunity to develop more potent, biologically active DDK inhibitors for future evaluation. Proteolysis of key regulatory factors is an important control element of gene activity both in eukaryotic and prokaryotic cells. In bacteria degradation by ATP-dependent proteases, belonging to the AAA+ superfamily, participates in regulation of many developmental pathways: the heat shock response, starvation adaptation, DNA damage repair, capsular polysaccharide biosynthesis, sporulation and control of bacteriophage development. Specific adaptor proteins are known to modify the interaction of substrates with ATP-dependent proteases. However, there are only three known intracellular inhibitory polypeptides. The phage T4 PinA protein
results highlight the cross-reactivity of several kinase inhibitors with DDK and also reveal an opportunity to develop more potent, biologically active DDK inhibitors for future evaluation. Proteolysis of key regulatory factors is an important control element of gene activity both in eukaryotic and prokaryotic cells. In bacteria degradation by ATP-dependent proteases, belonging to the AAA+ superfamily, participates in regulation of many developmental pathways: the heat shock response, starvation adaptation, DNA damage repair, capsular polysaccharide biosynthesis, sporulation and control of bacteriophage development. Specific adaptor proteins are known to modify the interaction of substrates with ATP-dependent proteases. However, there are only three known intracellular inhibitory polypeptides. The phage T4 PinA protein 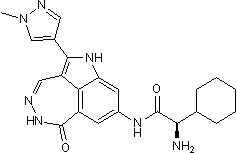 drug screen using the KIN001 library. Our study also highlighted the potential of Jak inhibitors to synergize with PKC412 and AC220 as well as enhance their apoptotic activity against mutant FLT3-expressing cells cultured in the presence of SCM. While the significance of stromal-derived growth factors in viability enhancement and cytoprotection of leukemic stem cells cannot be denied, not all hematologic malignancies can be rescued from programmed cell death by secreted cytokines in the absence of direct communication with the stromal cells themselves. As examples, protection of AML cells and B-lineage ALL cells from spontaneous and/or drug-induced apoptosis was observed to depend on direct bone marrow fibroblast cell:leukemic cell interaction. Similarly, protection of CLL cells from apoptosis depends on adherence of these cells to bone marrow stromal layers, and adhesion between bone marrow stroma and myeloma cells is necessary for protection of these cells from drug-induced apoptosis. Thus, the direct interaction between stromal cells and leukemic cells is important to fully
drug screen using the KIN001 library. Our study also highlighted the potential of Jak inhibitors to synergize with PKC412 and AC220 as well as enhance their apoptotic activity against mutant FLT3-expressing cells cultured in the presence of SCM. While the significance of stromal-derived growth factors in viability enhancement and cytoprotection of leukemic stem cells cannot be denied, not all hematologic malignancies can be rescued from programmed cell death by secreted cytokines in the absence of direct communication with the stromal cells themselves. As examples, protection of AML cells and B-lineage ALL cells from spontaneous and/or drug-induced apoptosis was observed to depend on direct bone marrow fibroblast cell:leukemic cell interaction. Similarly, protection of CLL cells from apoptosis depends on adherence of these cells to bone marrow stromal layers, and adhesion between bone marrow stroma and myeloma cells is necessary for protection of these cells from drug-induced apoptosis. Thus, the direct interaction between stromal cells and leukemic cells is important to fully