To investigate why tau phosphorylation at Ser199, Ser202, Ser396 and Ser422 was SCH772984 increased when O-GlcNAcylation was elevated upon thiametG treatment, we studied the major tau kinases. We first studied GSK-3b and its upstream regulating pathway, the PI3K-AKT signaling pathway. Which in turn 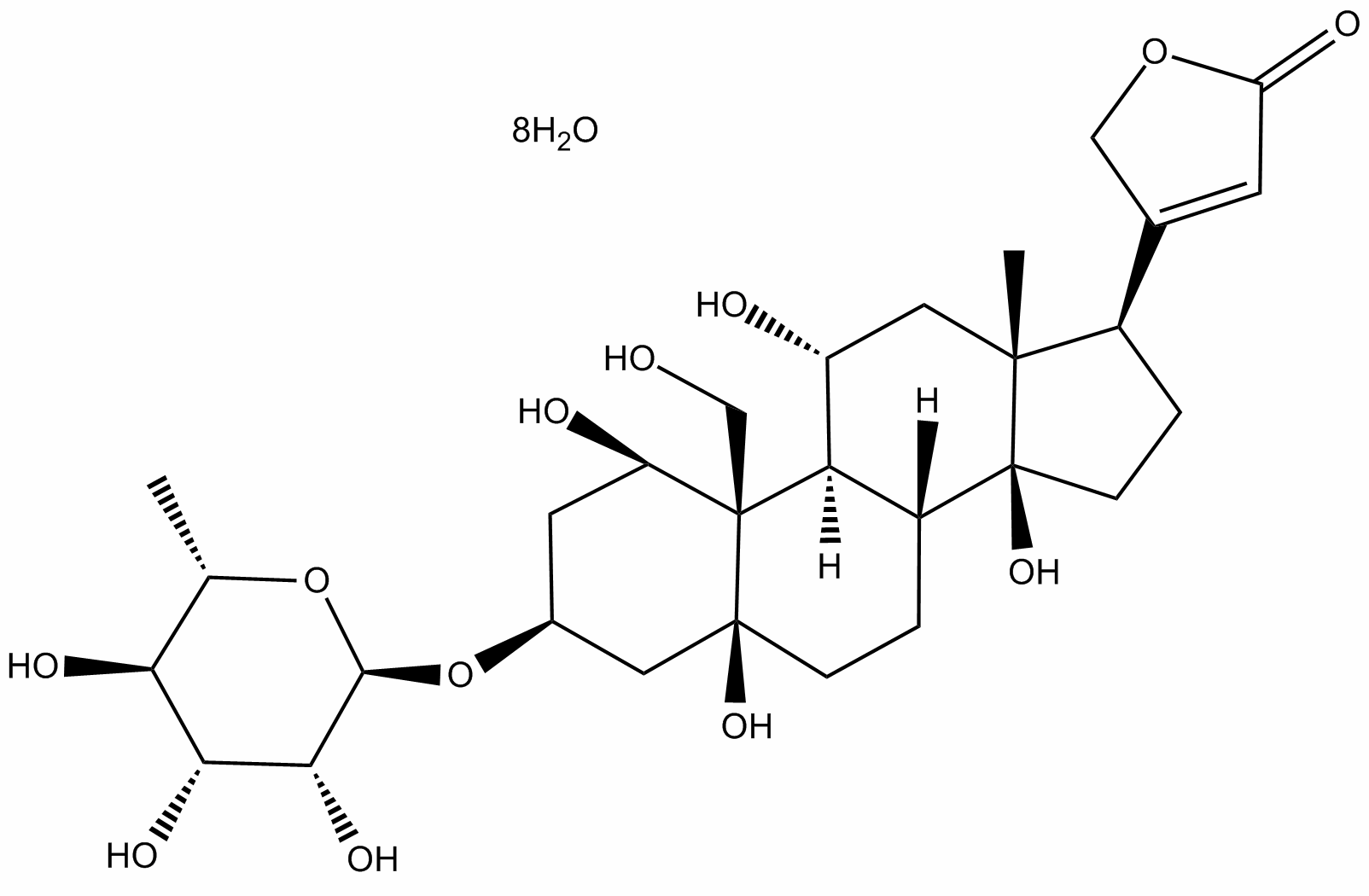 is regulated by PI3K and other factors. We found that thiamet-G treatment did not significantly alter the level of GSK-3b except after treatment for 9�C24 h, but blocked its phosphorylation at Ser9 completely, suggesting that GSK-3b was markedly activated under these conditions. Phosphorylation of GSK-3b at Tyr216, which makes it more active, was also increased 24 h after thiamet-G treatment. Consistent with the almost complete dephosphorylation of GSK-3b at Ser9, Ser473 and Thr308 phosphorylation of AKT, which determines its kinase activity, was also blocked by thiamet-G treatment. However, we did not find any significant changes of either the level or the activation of PI3K, the major upstream kinase of AKT, in the mouse brain after thiamet-G treatment. These results suggest that thiamet-G induced over-activation of GSK-3b via inhibition of AKT phosphorylation. The over-activation of GSK-3b may explain the increased tau phosphorylation at Ser199, Ser202, Ser396 and Ser422, NVP-BKM120 because these sites are the phosphorylation sites catalyzed mainly by GSK-3b. CDK5 is the second most important tau kinase in the brainand is activated by p35/p25. We thus also studied the level of CDK5 and its activators. We found that thiamet-G did not alter either CDK5 or p35. P25, which is the truncated and a more active form of p35, was not detectable in the mouse brain. To elucidate the complex regulation of site-specific phosphorylation induced by thiamet-G, we employed cultured cells, because cell cultures are more easily manipulated. We first selected AHP cells because they are more close to brain neurons than tumor cell lines. As expected, treatment of AHP cells with 20 nM thiamet-G increased protein O-GlcNAcylation. While we observed decreased tau phosphorylation at several phosphorylation sites in the AHP cells after thiamet-G treatment, it did not induce any significant increase in tau phosphorylation at the phosphorylation sites studied except at a transient elevation of Ser396 at 30 min after the treatment. We then investigated the levels and the activation of GSK-3b and the upstream PI3K-AKT signaling transduction pathway, as well as CDK5/p35. We did not find any significant changes upon the treatments with thiamet-G in AHP cells. These results further support our conclusion above that the increased tau phosphorylation at some sites observed in the thiamet-G treated mouse brains was due to GSK3b activation. To learn whether the phenomena we observed in undifferentiated AHP cells were specific to these cells, we also performed similar experiments in differentiated AHP cells and differentiated PC12 cells. As seen in proliferating AHP cells, we did not observe any marked elevation of tau phosphorylation at any phosphorylation sites or changes of tau kinases upon thiamet-G treatments in these two types of cells. Thiamet-G is a highly specific OGA inhibitor that was synthesized based on rationale design. Initial studies indicated that this compound reduce tau phosphorylation at some phosphorylation sites that can be abnormally phosphorylated in AD, suggesting that OGA inhibition may offer a potential therapeutic approach for slowing tau-mediated neurodegeneration seen in AD and other tauopathies. Because tau phosphorylation at different sites has different impacts on tau’s function and pathology, investigating the role of thiamet-G on site-specific tau phosphorylation is needed.
is regulated by PI3K and other factors. We found that thiamet-G treatment did not significantly alter the level of GSK-3b except after treatment for 9�C24 h, but blocked its phosphorylation at Ser9 completely, suggesting that GSK-3b was markedly activated under these conditions. Phosphorylation of GSK-3b at Tyr216, which makes it more active, was also increased 24 h after thiamet-G treatment. Consistent with the almost complete dephosphorylation of GSK-3b at Ser9, Ser473 and Thr308 phosphorylation of AKT, which determines its kinase activity, was also blocked by thiamet-G treatment. However, we did not find any significant changes of either the level or the activation of PI3K, the major upstream kinase of AKT, in the mouse brain after thiamet-G treatment. These results suggest that thiamet-G induced over-activation of GSK-3b via inhibition of AKT phosphorylation. The over-activation of GSK-3b may explain the increased tau phosphorylation at Ser199, Ser202, Ser396 and Ser422, NVP-BKM120 because these sites are the phosphorylation sites catalyzed mainly by GSK-3b. CDK5 is the second most important tau kinase in the brainand is activated by p35/p25. We thus also studied the level of CDK5 and its activators. We found that thiamet-G did not alter either CDK5 or p35. P25, which is the truncated and a more active form of p35, was not detectable in the mouse brain. To elucidate the complex regulation of site-specific phosphorylation induced by thiamet-G, we employed cultured cells, because cell cultures are more easily manipulated. We first selected AHP cells because they are more close to brain neurons than tumor cell lines. As expected, treatment of AHP cells with 20 nM thiamet-G increased protein O-GlcNAcylation. While we observed decreased tau phosphorylation at several phosphorylation sites in the AHP cells after thiamet-G treatment, it did not induce any significant increase in tau phosphorylation at the phosphorylation sites studied except at a transient elevation of Ser396 at 30 min after the treatment. We then investigated the levels and the activation of GSK-3b and the upstream PI3K-AKT signaling transduction pathway, as well as CDK5/p35. We did not find any significant changes upon the treatments with thiamet-G in AHP cells. These results further support our conclusion above that the increased tau phosphorylation at some sites observed in the thiamet-G treated mouse brains was due to GSK3b activation. To learn whether the phenomena we observed in undifferentiated AHP cells were specific to these cells, we also performed similar experiments in differentiated AHP cells and differentiated PC12 cells. As seen in proliferating AHP cells, we did not observe any marked elevation of tau phosphorylation at any phosphorylation sites or changes of tau kinases upon thiamet-G treatments in these two types of cells. Thiamet-G is a highly specific OGA inhibitor that was synthesized based on rationale design. Initial studies indicated that this compound reduce tau phosphorylation at some phosphorylation sites that can be abnormally phosphorylated in AD, suggesting that OGA inhibition may offer a potential therapeutic approach for slowing tau-mediated neurodegeneration seen in AD and other tauopathies. Because tau phosphorylation at different sites has different impacts on tau’s function and pathology, investigating the role of thiamet-G on site-specific tau phosphorylation is needed.
GSK-3b activity is mainly regulated negatively via its phosphorylation at Ser9 by AKT
Leave a reply
 LMP7 is critical for proteasome activity, and it was recently reported that LMP7deficiency is associated with reduced severity of DSS-induced colitis in mice. Patients with inflammatory bowel disease exhibit high levels of LMP7 in the inflamed gut, and their increased proteasome activity induced by high levels of expression of immunoproteasome subunits mediates sustained activation of NF-kB. These results suggest that proteasome inhibitors may ameliorate inflammatory bowel disease. In this study, bortezomib administration substantially reduced the severity of DSS-induced colitis as well as significantly enhancing apoptosis and IkB expression of CD4 + and CD8 + T cells during DSS-induced colitis. Thus, NF-kB inhibition is likely to contribute to bortezomib-induced cell death in T cells, thereby suppressing DSS-induced colitis. Bortezomib treatment of mice with lupus-like disease significantly improves the disease severity by reducing the numbers of both CD4 + and CD8 + T cells in the spleen. Bortezomib treatment also demonstrates significant protection from acute graft-versus-host disease in a murine allogeneic bone marrow transplantation model by inhibiting allogeneic T cell proliferation. By contrast, bortezomib administration largely eliminates plasma cells but not T cells or B cells in murine models of human systemic lupus erythematosus. In the current study, bortezomib treatment reduced the numbers of CD4 + and CD8 + T cells, but not B cells or macrophages during DSS-induced colitis. It has been reported that proliferating T cells are more sensitive to
LMP7 is critical for proteasome activity, and it was recently reported that LMP7deficiency is associated with reduced severity of DSS-induced colitis in mice. Patients with inflammatory bowel disease exhibit high levels of LMP7 in the inflamed gut, and their increased proteasome activity induced by high levels of expression of immunoproteasome subunits mediates sustained activation of NF-kB. These results suggest that proteasome inhibitors may ameliorate inflammatory bowel disease. In this study, bortezomib administration substantially reduced the severity of DSS-induced colitis as well as significantly enhancing apoptosis and IkB expression of CD4 + and CD8 + T cells during DSS-induced colitis. Thus, NF-kB inhibition is likely to contribute to bortezomib-induced cell death in T cells, thereby suppressing DSS-induced colitis. Bortezomib treatment of mice with lupus-like disease significantly improves the disease severity by reducing the numbers of both CD4 + and CD8 + T cells in the spleen. Bortezomib treatment also demonstrates significant protection from acute graft-versus-host disease in a murine allogeneic bone marrow transplantation model by inhibiting allogeneic T cell proliferation. By contrast, bortezomib administration largely eliminates plasma cells but not T cells or B cells in murine models of human systemic lupus erythematosus. In the current study, bortezomib treatment reduced the numbers of CD4 + and CD8 + T cells, but not B cells or macrophages during DSS-induced colitis. It has been reported that proliferating T cells are more sensitive to  tumor cell lines, including breast cancer cells with mutant or amplified PIK3CA. BEZ235 showed antitumor activity in nude mice with few side effects. A recent report from a phase I study of BEZ235 in 59 patients with advanced solid tumors demonstrated antitumor effects and a favorable safety profile. ZSTK474, a pan-class I PI3K inhibitor, also demonstrated high potency against a panel of cancer cell lines and human tumor xenografts without toxicity to major organs. As discussed above, among all drugs tested, the agents which produced synergy with temsirolimus in our models were BEZ235 and ZSTK474. A main conclusion of our study is that combination treatment of ZSTK474 or BEZ235 with temsirolimus synergizes to decrease
tumor cell lines, including breast cancer cells with mutant or amplified PIK3CA. BEZ235 showed antitumor activity in nude mice with few side effects. A recent report from a phase I study of BEZ235 in 59 patients with advanced solid tumors demonstrated antitumor effects and a favorable safety profile. ZSTK474, a pan-class I PI3K inhibitor, also demonstrated high potency against a panel of cancer cell lines and human tumor xenografts without toxicity to major organs. As discussed above, among all drugs tested, the agents which produced synergy with temsirolimus in our models were BEZ235 and ZSTK474. A main conclusion of our study is that combination treatment of ZSTK474 or BEZ235 with temsirolimus synergizes to decrease  the polymerase enzyme. A previous study revealed high frequency of alterations of DSB repair genes in MSI positive ECs, where MRE11 and RAD50 exhibited heterozygous and homozygous mutations in 51% and 17%, respectively, without examining the impact of the loss of the proteins. Mutations of the MRE11 polyT allele are predominantly heterozygous mutations and it has been suggested that only those with mutations of two and more nucleotides as well as homozygous mutations have a functional impact in terms of loss of function. In this report, we cannot confirm the high frequency of intronic mutations but provide evidence that MRE11 protein is lost in a substantial proportion of ECs. Recently, it has been shown that whole exon sequencing of MRE11 revealed mutations in 1.9% of the EC tumours within the exons. However, intronic mutations have not been assessed, explaining why the frequency of MRE11 mutations is reported to be low not only in the study by Price et al. but also in a recent one by The Cancer Genome Atlas Research Network. PARP inhibitors have shown remarkable sensitivity in BRCA1/ 2-deficient tumour models in vitro as well as in clinical trials involving
the polymerase enzyme. A previous study revealed high frequency of alterations of DSB repair genes in MSI positive ECs, where MRE11 and RAD50 exhibited heterozygous and homozygous mutations in 51% and 17%, respectively, without examining the impact of the loss of the proteins. Mutations of the MRE11 polyT allele are predominantly heterozygous mutations and it has been suggested that only those with mutations of two and more nucleotides as well as homozygous mutations have a functional impact in terms of loss of function. In this report, we cannot confirm the high frequency of intronic mutations but provide evidence that MRE11 protein is lost in a substantial proportion of ECs. Recently, it has been shown that whole exon sequencing of MRE11 revealed mutations in 1.9% of the EC tumours within the exons. However, intronic mutations have not been assessed, explaining why the frequency of MRE11 mutations is reported to be low not only in the study by Price et al. but also in a recent one by The Cancer Genome Atlas Research Network. PARP inhibitors have shown remarkable sensitivity in BRCA1/ 2-deficient tumour models in vitro as well as in clinical trials involving  We found that all molecules previously reported to bind to c-MYC also bound to MYCN. Treatment with the small molecules furthermore interfered with the MYCN/ MAX interaction and caused protein degradation, apoptosis, differentiation and lipid formation to different extents in MYCNamplified NB cells. To date several research
We found that all molecules previously reported to bind to c-MYC also bound to MYCN. Treatment with the small molecules furthermore interfered with the MYCN/ MAX interaction and caused protein degradation, apoptosis, differentiation and lipid formation to different extents in MYCNamplified NB cells. To date several research