The mutational analysis from the static structure normally ignores short or long range conformational changes and they do not include the dynamic effects caused by thermal motions. The molecular dynamics simulations and molecular mechanics-Poisson-Boltzmann surface area calculations on the problem of imatinib resistance by various BCR-ABL mutations has been studied by Lee et al.,. Computational simulations can provide atomic level description of structural details, energy landscape, dynamic behaviours, and other properties which are difficult to be obtained from the experimental studies. Here, we report the MD simulations, solvated interaction energies free energy calculations of ponatinib with native and mutants of BCR-ABL kinase. We have also calculated the contributions from individual amino acid BEZ235 residues in the active site of all complexes to provide the molecular basis for inhibition. To our knowledge these Reversine studies have not been carried out before and our results provide detailed information about the molecular mechanisms of inhibition of native and various mutant BCR-ABL tyrosine kinases when bound to ponatinib. The native and mutant ABL kinase �C ponatinib complexes with explicit water molecules and sodium ions for charge neutralization were subjected to 25 ns MD simulations. The fourteen BCR-ABL mutants studied in this work collectively represent more than 95% of clinically observed mutations that are imatinib resistant. With the exception of T315I, most BCR-ABL mutations are inhibited by dasatinib and nilotinib. Ponatinib inhibits native and all mutant ABL kinases with high affinity, although some mutants have slightly greater inhibition than the others. The ATP competitive inhibitors of ABL kinase are classified into DFG-in or DFG-out classes depending on their binding interactions with kinase domain. Ponatinib binds to ABL kinase domain with a DFG-out conformation and serves to distribute binding energy over a wide range of amino acid residues in the active site as shown in Figure 1. The presence of such optimized and distributed binding interactions has the potential to allow ponatinib to withstand modest reduction in potency caused by single mutation. For our convenience; we grouped these mutations by the region of their location in ABL kinase structure. These regions include the P-loop mutants M244V, G250E, Q252H, Y253F, Y253H, E255K, and E255V; gatekeeper residue mutants T315A and T315I; hinge region mutants F317L and F317V; activation loop mutant H396P and other mutants M351T and F359V. The location of mutations in BCR-ABL kinase is shown in Figure 2. In the ABL kinase, amino acid residues Tyr253, Thr315, Phe317 and Phe359 are located in close contact with ponatinib and therefore affect the binding and activity of inhibitor. The Ploop mutant residues Gly250, Gln252 and Glu255 are not in direct contact with ponatinib, but share non-bonding interactions with inhibitor. The rest of the mutations Met244, Met351 and His396 are located away from inhibitor binding site, but intriguingly display ponatinib based inhibition. SIE calculations from MD trajectories measure the free energy of complex formation. Table 1 shows the calculated free energies for native and 14 mutant BCR-ABL �C ponatinib complexes. The intermolecular vdW, intermolecular coulomb and change in surface area are shown in Table 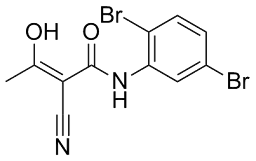 1. This table indicates that IC50 values vary from 0.5 nM to 36 nM and SIE values calculated from this work are in the range 210.03 kcal/mol to 210.67 kcal/mol. Though there is no direct correlation between IC50 and SIE values, it can be observed that their respective values lie within a narrow range. Many patients eventually developed imatinib resistance, usually associated with above mentioned mutations in ABL kinase domain that either directly or indirectly effects the binding affinity of imatinib to ABL. The most common gatekeeper residue mutation T315I that accounts for 15�C20% of clinically observed mutations is completely resistant to imatinib, nilotinib and dasatinib. Native and T315I BCR-ABL kinases complexed with dasatinib are subjected to 25 ns of MD simulations and SIE binding free energies are calculated. The analysis of dasatinib complexed with native and T315I mutant BCR-ABL kinases revealed that native complex has relatively higher SIE free energy than when complexed with T315I that signifies the greater affinity of dasatinib for native compared to mutant BCR-ABL kinase.
1. This table indicates that IC50 values vary from 0.5 nM to 36 nM and SIE values calculated from this work are in the range 210.03 kcal/mol to 210.67 kcal/mol. Though there is no direct correlation between IC50 and SIE values, it can be observed that their respective values lie within a narrow range. Many patients eventually developed imatinib resistance, usually associated with above mentioned mutations in ABL kinase domain that either directly or indirectly effects the binding affinity of imatinib to ABL. The most common gatekeeper residue mutation T315I that accounts for 15�C20% of clinically observed mutations is completely resistant to imatinib, nilotinib and dasatinib. Native and T315I BCR-ABL kinases complexed with dasatinib are subjected to 25 ns of MD simulations and SIE binding free energies are calculated. The analysis of dasatinib complexed with native and T315I mutant BCR-ABL kinases revealed that native complex has relatively higher SIE free energy than when complexed with T315I that signifies the greater affinity of dasatinib for native compared to mutant BCR-ABL kinase.
The RMSD of BCR-ABL kinase ponatinib complexes shown indicated that in the native complex
Leave a reply
 equally well. Therefore, it is still of great interest to find new types of inhibitors. In order to facilitate this search, various screening methods have been employed so far. All of these have relied on monitoring the cleavage of a substrate through gel-based, FRET or MALDI mass spectrometry techniques. However, a limitation of these methods is the availability of a matching protein or polypeptide substrate. Rhomboids from one species may cleave substrates from another species, but this is not a general rule. We therefore reasoned that it would be beneficial to develop an inhibitor assay for rhomboid proteases that does not rely on a substrate at all. A few years ago Cravatt and co-workers developed a highthroughput inhibitor screening method that uses fluorescent activity-based probes. ABPs are small molecules that covalently bind to the active form of an enzyme, but not to an inactivated or zymogen form. ABPs generally consist of a tag, a spacer and an electrophilic group that traps an active site nucleophile. The binding event can be detected by a variety of techniques, such as gel-scanning, biotin blot or fluorescent microscopy, depending on the tagging moiety. When appended to a fluorescent dye, the binding of an ABP can be detected by fluorescence polarization. This so-called fluorescence polarization activity-based protein profiling has been used in inhibitor high-throughput screens for a variety of poorly characterized enzymes.
equally well. Therefore, it is still of great interest to find new types of inhibitors. In order to facilitate this search, various screening methods have been employed so far. All of these have relied on monitoring the cleavage of a substrate through gel-based, FRET or MALDI mass spectrometry techniques. However, a limitation of these methods is the availability of a matching protein or polypeptide substrate. Rhomboids from one species may cleave substrates from another species, but this is not a general rule. We therefore reasoned that it would be beneficial to develop an inhibitor assay for rhomboid proteases that does not rely on a substrate at all. A few years ago Cravatt and co-workers developed a highthroughput inhibitor screening method that uses fluorescent activity-based probes. ABPs are small molecules that covalently bind to the active form of an enzyme, but not to an inactivated or zymogen form. ABPs generally consist of a tag, a spacer and an electrophilic group that traps an active site nucleophile. The binding event can be detected by a variety of techniques, such as gel-scanning, biotin blot or fluorescent microscopy, depending on the tagging moiety. When appended to a fluorescent dye, the binding of an ABP can be detected by fluorescence polarization. This so-called fluorescence polarization activity-based protein profiling has been used in inhibitor high-throughput screens for a variety of poorly characterized enzymes. and in the case of lysosomes ‘lysosomotropism’. This accumulation of desipramine 168 results in detachment of the ASM from the inner lysosomal membraneand its subsequent inactivation, probably by proteolytic degradation. Weak bases, therefore, do not directly inhibit ASM, but result in a functional inhibition of ASM. We have thus proposed the acronym FIASMA for Functional Inhibitor of Acid SphingoMyelinAse. According to this model, functional inhibition of ASM requires high lysosomal concentrations of a weak basic drug. Previously, we have shown that functional inhibition of ASM is related to high pKa- and high logP-values and have characterized several new FIASMAs, including the antidepressant drugs doxepine 63, fluoxetine 104, maprotilin 109, nortriptyline 114, paroxetine 118 and sertraline 124. The aims of the present study wereto identify more FIASMAs,to further improve the in silico prediction of functional ASM inhibition by developing compact and easily-interpretable models with high internal
and in the case of lysosomes ‘lysosomotropism’. This accumulation of desipramine 168 results in detachment of the ASM from the inner lysosomal membraneand its subsequent inactivation, probably by proteolytic degradation. Weak bases, therefore, do not directly inhibit ASM, but result in a functional inhibition of ASM. We have thus proposed the acronym FIASMA for Functional Inhibitor of Acid SphingoMyelinAse. According to this model, functional inhibition of ASM requires high lysosomal concentrations of a weak basic drug. Previously, we have shown that functional inhibition of ASM is related to high pKa- and high logP-values and have characterized several new FIASMAs, including the antidepressant drugs doxepine 63, fluoxetine 104, maprotilin 109, nortriptyline 114, paroxetine 118 and sertraline 124. The aims of the present study wereto identify more FIASMAs,to further improve the in silico prediction of functional ASM inhibition by developing compact and easily-interpretable models with high internal  CDE. The expression of mutants of proteins involved in the clathrin machinery, such as Dynamin2-K44A, the carboxy terminus of AP180, and clathrin hubs, has proven quite effective. More recently siRNA-mediated depletion of the clathrin heavy chain, subunits of the AP2 adaptor, and dynamin 2have abolished CDE in cells. The drawback of these genetic approaches is that they require days to take effect and may lead to many indirect effects or compensatory cellular responses that make interpretation of the findings sometimes difficult. Use of a number of acute cellular treatments including cytosol acidification and hypotonic treatment can be effective at blocking endocytosis of CDE cargobut these treatments are non-specific and may also affect CIE. Recently, new compounds that selectively target proteins involved in CDE have been identified with the promise that these could be used to acutely inhibit this process. These include compounds that specifically target dynamin such as dynasoreand the dynoles. Since dynamin is required for all forms of CDE and is used in some forms of CIE, a compound that selectively targets clathrin was developed by Haucke and colleagues. This compound, named pitstop 2, was designed and shown to bind to and block interactions between the amino terminal domain of clathrin heavy chain and amphiphysin, one of many proteins shown to bind to this domain of clathrin. In cells, pitstop 2 was shown to inhibit endocytosis of transferrin receptor, a CDE cargo
CDE. The expression of mutants of proteins involved in the clathrin machinery, such as Dynamin2-K44A, the carboxy terminus of AP180, and clathrin hubs, has proven quite effective. More recently siRNA-mediated depletion of the clathrin heavy chain, subunits of the AP2 adaptor, and dynamin 2have abolished CDE in cells. The drawback of these genetic approaches is that they require days to take effect and may lead to many indirect effects or compensatory cellular responses that make interpretation of the findings sometimes difficult. Use of a number of acute cellular treatments including cytosol acidification and hypotonic treatment can be effective at blocking endocytosis of CDE cargobut these treatments are non-specific and may also affect CIE. Recently, new compounds that selectively target proteins involved in CDE have been identified with the promise that these could be used to acutely inhibit this process. These include compounds that specifically target dynamin such as dynasoreand the dynoles. Since dynamin is required for all forms of CDE and is used in some forms of CIE, a compound that selectively targets clathrin was developed by Haucke and colleagues. This compound, named pitstop 2, was designed and shown to bind to and block interactions between the amino terminal domain of clathrin heavy chain and amphiphysin, one of many proteins shown to bind to this domain of clathrin. In cells, pitstop 2 was shown to inhibit endocytosis of transferrin receptor, a CDE cargo  inhibitor when used at high doses. Phosphorylation of these kinases has been reported to be regulated negatively by O-GlcNAcylation. It is worth noting that different effects of OGA inhibition on phosphorylation of AKT and GSK-3 have been reported. Elevation of O-GlcNAcylation in skeletal muscles after OGA inhibition using another inhibitor, PUGNAc, does not significantly alter insulin-stimulated phosphorylation of AKT or GSK-3. In differentiated 3T3-L1 adipocytes, two different OGA inhibitors have been found to increase O-GlcNAc levels but not alter insulin-stimulated phosphorylation of AKT nor induce insulin resistance either. Therefore, it remains somewhat unclear as to the effects of OGA inhibition on alteration of GSK3b levels and AKT activity; the effects observed here could stem from high-dose inhibition of OGA, or alternatively from off-target effects of using the inhibitor at a high dose. Tau is abnormally hyperphosphorylated and aggregated in AD and other tauopathies. Previous studies from our and other groups have demonstrated differential roles of tau phosphorylation at various phosphorylation sites. A quantitative in vitro study demonstrated that phosphorylation of tau at Ser262, Thr231, and Ser235 inhibits its binding to microtubules by,35%,,25%, and,10%, respectively. In vitro kinetic studies of the binding between hyperphosphorylated tau and normal tau suggest that Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/ Ser356 and Ser422 are among the critical phosphorylation sites that convert tau to an inhibitory molecule that sequesters normal microtubule-associated proteins from microtubules. Further phosphorylation at Thr231, Ser396, and Ser422 promotes selfaggregation of tau into filaments. It is obvious that tau phosphorylation at various sites impacts tau activity and aggregation collectively. Our recent study has demonstrated that tau phosphorylation at the proline-rich region, which is located upstream of the microtubule-binding domains, inhibits its microtubule
inhibitor when used at high doses. Phosphorylation of these kinases has been reported to be regulated negatively by O-GlcNAcylation. It is worth noting that different effects of OGA inhibition on phosphorylation of AKT and GSK-3 have been reported. Elevation of O-GlcNAcylation in skeletal muscles after OGA inhibition using another inhibitor, PUGNAc, does not significantly alter insulin-stimulated phosphorylation of AKT or GSK-3. In differentiated 3T3-L1 adipocytes, two different OGA inhibitors have been found to increase O-GlcNAc levels but not alter insulin-stimulated phosphorylation of AKT nor induce insulin resistance either. Therefore, it remains somewhat unclear as to the effects of OGA inhibition on alteration of GSK3b levels and AKT activity; the effects observed here could stem from high-dose inhibition of OGA, or alternatively from off-target effects of using the inhibitor at a high dose. Tau is abnormally hyperphosphorylated and aggregated in AD and other tauopathies. Previous studies from our and other groups have demonstrated differential roles of tau phosphorylation at various phosphorylation sites. A quantitative in vitro study demonstrated that phosphorylation of tau at Ser262, Thr231, and Ser235 inhibits its binding to microtubules by,35%,,25%, and,10%, respectively. In vitro kinetic studies of the binding between hyperphosphorylated tau and normal tau suggest that Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/ Ser356 and Ser422 are among the critical phosphorylation sites that convert tau to an inhibitory molecule that sequesters normal microtubule-associated proteins from microtubules. Further phosphorylation at Thr231, Ser396, and Ser422 promotes selfaggregation of tau into filaments. It is obvious that tau phosphorylation at various sites impacts tau activity and aggregation collectively. Our recent study has demonstrated that tau phosphorylation at the proline-rich region, which is located upstream of the microtubule-binding domains, inhibits its microtubule