If the same idea was used in gene selection, then the selected signature gene lists would be similar for different studies. Thus, there is a strong possibility that gene selections were influenced by variations in sample collection, sample size, data processing, and microarray platform. Our method does not use survival time as a parameter for gene selection; rather, it used a gene clustering approach instead of group statistics. It is not unexpected that our gene list does not overlap previously reported lung cancer signature genes as our signature development approach is quite different. The ratio of 3,4,5-Trimethoxyphenylacetic acid two-gene expression within an individual patient has been reported as a biomarker signature development in lung cancer diagnosis and prognosis as well as for breast cancer prognosis. The single two-gene ratio or geometric mean of several two-gene ratios was selected between the treatment failures and the treatment responders from the training data samples. The single two-gene ratio works well for cancer cell type classification or diagnosis; for example, between malignant pleural mesothelioma and adenocarcinoma, but it may not be able to reflect the complex tumor progression process for prognosis. In some cases, there could be substantial variation of the two genes among different samples. Therefore 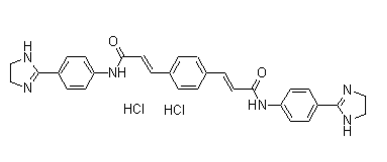 many new studies in recent years still rely on the Cox regression modeling to build the prognostic signatures. Most of these models applied the gene expression value to the Cox proportional coefficient of each signature gene and combined them as the patient risk scores. Some models computed the probability of a patient falling into the low-risk or high-risk class as the patient risk scores. However, there are difficulties in using overall survival as an endpoint in prognostic modeling in cancer. The expression variations of the same gene among individual subjects are substantial. Some genes associated with other aggressive diseases may be present in a subject’s tumor. Similarly, a subject might develop and succumb to some other clinical condition shortly after diagnosis. In these instances, no correlation exists between gene expression and subject survival. The complex models could learn the expression variable as well as other variations precisely, which would result in low reproducibility if used for a different data set. Instead, we return to the two-group gene expression ratio approach but select these two groups of genes using differences between normal lung cells and lung cancer cells that represent the whole Yin and Yang effects of the cell. Among the Yin genes we selected included pathways and networks connected to the canonical Molecular Mechanisms of Cancer pathway. The Yang genes are connected to the Retinoic acid receptor activation pathway and the Hepatic Stellate Cell Activation pathway. RAR activation induces cell differentiation and may antagonize cancer progression because retinoic acid or vitamin A is required for the differentiated state of normal cells. Hepatic Stellate Cells play a key role in the storage and transport of retinoids and the lung tissue harbors the Hepatic Stellate-like cells. These two pathways alter the balance between Yin and Yang, consequently altering patient survival. A useful prognostic signature should not only predict the patient’s prognosis, but should also help clinical therapeutic Salvianolic-acid-B decision making. Although surgery is a standard treatment for early stage lung cancer, more than 20% of stage I patients will relapse. This cohort of patients may benefit from chemotherapy.
many new studies in recent years still rely on the Cox regression modeling to build the prognostic signatures. Most of these models applied the gene expression value to the Cox proportional coefficient of each signature gene and combined them as the patient risk scores. Some models computed the probability of a patient falling into the low-risk or high-risk class as the patient risk scores. However, there are difficulties in using overall survival as an endpoint in prognostic modeling in cancer. The expression variations of the same gene among individual subjects are substantial. Some genes associated with other aggressive diseases may be present in a subject’s tumor. Similarly, a subject might develop and succumb to some other clinical condition shortly after diagnosis. In these instances, no correlation exists between gene expression and subject survival. The complex models could learn the expression variable as well as other variations precisely, which would result in low reproducibility if used for a different data set. Instead, we return to the two-group gene expression ratio approach but select these two groups of genes using differences between normal lung cells and lung cancer cells that represent the whole Yin and Yang effects of the cell. Among the Yin genes we selected included pathways and networks connected to the canonical Molecular Mechanisms of Cancer pathway. The Yang genes are connected to the Retinoic acid receptor activation pathway and the Hepatic Stellate Cell Activation pathway. RAR activation induces cell differentiation and may antagonize cancer progression because retinoic acid or vitamin A is required for the differentiated state of normal cells. Hepatic Stellate Cells play a key role in the storage and transport of retinoids and the lung tissue harbors the Hepatic Stellate-like cells. These two pathways alter the balance between Yin and Yang, consequently altering patient survival. A useful prognostic signature should not only predict the patient’s prognosis, but should also help clinical therapeutic Salvianolic-acid-B decision making. Although surgery is a standard treatment for early stage lung cancer, more than 20% of stage I patients will relapse. This cohort of patients may benefit from chemotherapy.
The published signatures showed little overlap in the genes identified as significant predictors of outcome
Leave a reply